3 Linux神器
视频课见 http://bioinfo.ke.qq.com。
3.1 正则表达式替换文本随心所欲
正则表达式 (regular expression)是用来做模糊匹配的,匹配符合特定模式的文本。最早来源于Unix系统中的sed和grep命令,在各个程序语言,如perl, python中也都有实现。不同程序语言中正则表达式语法大体通用,细节上又各自有自己的特色。

Figure 3.1: 正则表达式基本语法
# 假如有这么一个测试文件
ct@ehbio: ~/$ cat <<END >url.list
http://www.ehbio.com/ImageGP
http://www.ehbio.com/Training
http://www.ehbio.com/Esx
www.ehbio.com
http://www.ehbio.com/ImageGP/index.php/Home/Index/Lineplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/GOenrichmentplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/PHeatmap.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Boxplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Barplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Volcanoplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Manhattanplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Histogram.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/VennDiagram.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/UpsetView.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Densityplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/PCAplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/PCoAplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/CPCoAplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Sankey.html
http://blog.genesino.com
https://blog.csdn.net/qazplm12_3/
https://blog.csdn.net/woodcorpse/
blog.csdn.net/woodcorpse/article/details/79313846
ImageGP is one of the Best online plot.
123456789
END获取以https开头的行
ct@ehbio: ~/$ grep '^https' url.list
https://blog.csdn.net/qazplm12_3/
https://blog.csdn.net/woodcorpse/获取包含数字的行
ct@ehbio:~/$ grep '[0-9]' url.list
https://blog.csdn.net/qazplm12_3/
blog.csdn.net/woodcorpse/article/details/79313846
123456789获取空行
ct@ehbio:~/$ grep '^$' url.list
获取html结尾的行
ct@ehbio:~/$ grep 'html$' url.list
http://www.ehbio.com/ImageGP/index.php/Home/Index/Lineplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/GOenrichmentplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/PHeatmap.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Boxplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Barplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Volcanoplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Manhattanplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Histogram.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/VennDiagram.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/UpsetView.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Densityplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/PCAplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/PCoAplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/CPCoAplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Sankey.html获取Boxplot和Barplot的地址
# 未能满足要求
ct@ehbio:~/$ grep 'B.*plot' url.list
http://www.ehbio.com/ImageGP/index.php/Home/Index/Boxplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Barplot.html
ImageGP is one of the Best online plot.
# 一个办法:更长的匹配
ct@ehbio:~/$ grep 'B.*plot.html' url.list
http://www.ehbio.com/ImageGP/index.php/Home/Index/Boxplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Barplot.html
# 限定中间不能有空格
ct@ehbio:~/$ grep 'B[^ ]*plot' url.list
http://www.ehbio.com/ImageGP/index.php/Home/Index/Boxplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Barplot.html
# 限定中间只能有2个字符
ct@ehbio:~/$ grep 'B..plot' url.list
http://www.ehbio.com/ImageGP/index.php/Home/Index/Boxplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Barplot.html
# 2个字符的另外一种写法
ct@ehbio:~/$ grep -P 'B.{2}plot' url.list
http://www.ehbio.com/ImageGP/index.php/Home/Index/Boxplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Barplot.html
# 2个字符的再一种写法,只允许出现特定字符
ct@ehbio:~/$ grep -P 'B[arox]*plot' url.list
http://www.ehbio.com/ImageGP/index.php/Home/Index/Boxplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/Barplot.html获取PCA或PCoA相关的行
ct@ehbio:~/$ grep 'PCo*A' url.list
http://www.ehbio.com/ImageGP/index.php/Home/Index/PCAplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/PCoAplot.html
http://www.ehbio.com/ImageGP/index.php/Home/Index/CPCoAplot.html3.2 awk-生信分析不可缺少
前面的学习过程中已经提到了awk和sed的使用,作为一个引子。现在则详细列举关于awk常用的操作和一些偏门的操作。
3.2.1 awk基本参数解释
awk擅长于对文件按行操作,每次读取一行,然后进行相应的操作。
awk读取单个文件时的基本语法格式是awk 'BEGIN{OFS=FS="\t"}{print $0, $1;}' filename。
读取多个文件时的语法是awk 'BEGIN{OFS=FS="\t"}ARGIND==1{print $0, $1;}ARGIND==2{print $0;}' file1 file2。
awk后面的命令部分是用引号括起来的,可以单引号,可以双引号,但注意不能与内部命令中用到的引号相同,否则会导致最相邻的引号视为一组,引发解释错误。引号不可以嵌套
OFS: 文件输出时的列分隔符 (output field separtor)FS: 文件输入时的列分隔符 (field separtor)BEGIN: 设置初始参数,初始化变量END: 读完文件后做最终的处理其它
{}:循环读取文件的每一行$0表示一行内容;$1,$2, …$NF表示第一列,第二列到最后一列。NF (number of fields)文件多少列;NR (number of rows)文件读了多少行:FNR当前文件读了多少行,常用于多文件操作时。a[$1]=1: 索引操作,类似于python中的字典,在ID map,统计中有很多应用。
3.2.2 awk基本常见操作
- 针对特定列的计算,比如wig文件的标准化
# 注意除了第一行是空格,其它行都是tab键分割
ct@ehbio:~/sxbd$ cat <<END >ehbio.wig
variableStep chrom=chr2
300701 12
300702 10
300703 11
300704 13
300705 12.5
END
ct@localhost:~/sxbd$ awk 'BEGIN{OFS=FS="\t"}\
{if(FNR>1) $2=$2*10^6/(2.5*10^6); print $0}' ehbio.wig
variableStep chrom=chr2
300701 4.8
300702 4
300703 4.4
300704 5.2
300705 5- 计算某列内容出现的次数。
# 怎么获得count文件,应该不难吧
ct@ehbio:~/sxbd$ cat count
ID Type
Pou5f1 Pluripotency
Nanog Pluripotency
Sox2 Neuron
Tet1 Epigenetic
Tet3 Epigenetic
Myc Oncogene
ct@ehbio:~/sxbd$ awk 'BEGIN{OFS=FS="\t"}{if(FNR>1) a[$2]+=1;}END\
{print "Type\tCount"; for(i in a) print i,a[i];}' count
Type Count
Neuron 1
Epigenetic 2
Oncogene 1
Pluripotency 2
# 这个也可以用下面方式代替,但不直接
ct@ehbio:~/sxbd$ tail -n +2 count | cut -f 2 | sort | uniq -c | \
sed -e 's/^ *//' -e 's/ */\t/'
2 Epigenetic
1 Neuron
1 Oncogene
2 Pluripotency- 之前也提到过的列操作,从GTF文件中提取启动子区域
GRCh38.gtf可以从ftp://ftp.ensembl.org/pub/release-91/gtf/homo_sapiens/Homo_sapiens.GRCh38.91.gtf.gz下载,或使用提供的测试文件。
ct@ehbio:~/sxbd$ sed 's/"/\t/g' GRCh38.gtf | \
awk 'BEGIN{OFS=FS="\t"}{if($3=="gene") {ensn=$10; symbol=$16; \
if($7=="+") {start=$4-1; up=start-1000; if(up<0) up=0; dw=start+500; \
print $1,up, dw, ensn, symbol, $7;} else \
if($7=="-") {start=$5-1; up=start+1000; dw=start-500; \
if(dw<0) dw=0; print $1,dw,up,ensn,symbol,$7}}}' | sort -k1,1 -k2,2n \
>GRCh38.promoter.bed- 数据矩阵的格式化输出
ct@ehbio:~/sxbd$ cat numeric.matrix
ID A B C
a 1.002 1.234 1.999
b 2.333 4.232 0.889
ct@ehbio:~/sxbd$ awk '{if(FNR==1) print $0; \
else {printf "%s%s",$1,FS; for (i=2; i<=NF; i++) \
printf "%.1f %s", $i, (i==NF?RS:FS)}}' numeric.matrix
ID A B C
a 1.0 1.2 2.0
b 2.3 4.2 0.9 - 判断FASTQ文件中,输出质量值的长度是与序列长度不一致的序列ID
ct@ehbio:~/sxbd$ cat <<END | gzip -c >Test_2.fq.gz
>ehbio1
ACGTCGACGACGAGAGGAGAGGAGCCCTCTCGCCCGCCCTACTACCACCCACACACAACACAAGTGT
+
FFFFFFA$A#$$AFEEEEFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
>ehbio2
ACGTCGACGACGAGAGGAGAGGAGCCCTCTCGCCCGCCCTACTACCACCCACACACAACACAAGTGT
+
FFFFFF$A#$$AFEEEEFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
>ehbio3
ACGTCGACGACGAGAGGAGAGGAGCCTCTCGCCCGCCCTACTACCACCCACACACAACACAAGTGT
+
FFFFFFA$A#$$AFEEEEFFFFFFFFEFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
ENDct@ehbio:~/sxbd$ zcat Test_2.fq.gz | \
awk '{if(FNR%4==1) ID=$0; else if(FNR%4==2) seq_len=length($0); \
else if(FNR%4==0) {quality_len=length($0); if(seq_len!=quality_len) print ID; }}' - 筛选差异基因
# TAB键分割的文件
ct@ehbio:~/sxbd$ cat de_gene
ID log2fc padj
A 1 0.001
B -1 0.001
C 1 0.001
D 2 0.0001
E -0.51 0.051
F 0.1 0.1
G 1 0.1
ct@ehbio:~/sxbd$ awk '$3<0.05 || NR==1' de_gene
ID log2fc padj
A 1 0.001
B -1 0.001
C 1 0.001
D 2 0.0001
ct@ehbio:~/sxbd$ awk 'BEGIN{OFS=FS="\t"}{if(FNR==1) print $0; \
else {abs_log2fc=($2<0?$2*(-1):$2);if(abs_log2fc>=1 && $3<0.05) print $0;}}' de_gene
ID log2fc padj
A 1 0.001
B -1 0.001
C 1 0.001
D 2 0.0001- 筛选差异基因存储到不同的文件
ct@ehbio:~/sxbd$ awk 'BEGIN{OFS=FS="\t"; up="up"; dw="dw";}\
{if(FNR==1) {print $0 >up; print $0 >dw;} else \
if ($3<0.05) {if ($2>=1) print $0 >up; else if($2<=-1) print $0 >dw;}}' de_gene
ct@ehbio:~/sxbd$ head up dw
==> up <==
ID log2fc padj
A 1 0.001
C 1 0.001
D 2 0.0001
==> dw <==
ID log2fc padj
B -1 0.001- 筛选差异基因存储到不同的文件(自动版)
awk 'BEGIN{OFS=FS="\t"; up=ARGV[1]".up"; dw=ARGV[1]".dw";}\
{if(FNR==1) {print $0 >up; print $0 >dw;} \
else if ($3<=0.05) {if($2<=-1) print $0 >up; else if ($2>=1) print $0 >dw;}}' de_gene- ID map,常用于转换序列的ID、提取信息、合并信息等
# TAB键分割的文件
ct@ehbio:~/sxbd$ cat id_map
ENSM Symbol Entrez
ENSG00000280516 TMEM42 693149
ENSG00000281886 TGM4 7047
ENSG00000280873 DGKD 8527
ENSG00000281244 ADAMTS13 11093
ENSG00000280701 RP11-272D20.2
ENSG00000280674 ZDHHC3 51304
ENSG00000281623 Y_RNA
ENSG00000280479 CACFD1 11094
ENSG00000281165 SLC2A6 11182
ENSG00000281879 ABO 28
ENSG00000282873 BCL7A 605
ENSG00000280651 AC156455.1 100506691
ct@ehbio:~/sxbd$ vim ensm
ct@ehbio:~/sxbd$ cat ensm
ENSG00000281244
ENSG00000281165
ENSG00000282873
ct@ehbio:~/sxbd$ awk 'BEGIN{OFS=FS="\t"}ARGIND==1{if(FNR>1) ensm2entrez[$1]=$3;}\
ARGIND==2{print ensm2entrez[$1];}' id_map ensm
11093
11182
605
# 替代解决方案,注意 -w的使用,避免部分匹配。最稳妥的方式还是使用awk。
ct@ehbio:~/sxbd$ grep -w -f ensm id_map | cut -f 3
11093
11182
605- 转换大小写,
toupper,tolower
ct@ehbio:~/sxbd$ cat symbol
Tgm4
Dgkd
Abo
ct@ehbio:~/sxbd$ awk 'BEGIN{OFS=FS="\t"}ARGIND==1{if(FNR>1) ensm2entrez[$2]=$3;}\
ARGIND==2{print ensm2entrez[toupper($1)];}' id_map symbol
7047
8527
28- awk数值操作
ct@ehbio:~/sxbd$ cat <<END >file
2
4
3.1
4.5
5.4
7.6
8
END
# log2对数
ct@ehbio:~/sxbd$ awk 'BEGIN{OFS="\t";FS="\t"}{print log($0)/log(2)}' file
# 取整,四舍五入
ct@ehbio:~/sxbd$ awk 'BEGIN{OFS="\t";FS="\t"}{print int($1+0.5);}' file- awk定义函数
ct@ehbio:~/sxbd$ awk 'function abs(x){return ((x < 0.0) ? -x : x)}BEGIN{OFS="\t";FS="\t"}\
{pos[1]=$1;pos[2]=$2;pos[3]=$3;pos[4]=$4; len=asort(pos); \
for(i=len;i>1;i--) print abs(pos[i]-pos[i-1]);}' file- 字符串匹配
# TAB键分割的文件
ct@ehbio:~/sxbd$ cat ens.bed
1 100 105
2 100 105
3 100 105
Mt 100 105
X 100 105
ct@ehbio:~/sxbd$ awk 'BEGIN{OFS=FS="\t"}{if($1~/^[0-9XY]/) $1="chr"$1; else \
if($1~/M.*/) gsub(/M.*/, "chrM", $1); print $0}' ens.bed
chr1 100 105
chr2 100 105
chr3 100 105
chrM 100 105
chrX 100 105- 字符串分割
ct@ehbio:~/sxbd$ cat trinity_id
Trinity_C1_g1_i1
Trinity_C1_g1_i2
Trinity_C1_g1_i3
Trinity_C2_g1_i1
Trinity_C3_g1_i1
Trinity_C3_g3_i2
ct@ehbio:~/sxbd$ awk 'BEGIN{OFS=FS="\t"}{count=split($1, geneL, "_"); gene=geneL[1]; \
for(i=2;i<count;i++) gene=gene"_"geneL[i]; print gene,$1;}' trinity_id
Trinity_C1_g1 Trinity_C1_g1_i1
Trinity_C1_g1 Trinity_C1_g1_i2
Trinity_C1_g1 Trinity_C1_g1_i3
Trinity_C2_g1 Trinity_C2_g1_i1
Trinity_C3_g1 Trinity_C3_g1_i1
Trinity_C3_g3 Trinity_C3_g3_i2- awk脚本
ct@ehbio:~/sxbd$ cat <<END >grade.awk
BEGIN{OFS=FS="\t"; up=ARGV[1]".up"; dw=ARGV[1]".dw";}
{if(FNR==1) {print $0 >up; print $0 >dw;}
else if ($3<=0.05) {
if($2<=-1) print $0 >up;
else if ($2>=1) print $0 >dw;}
}
END
ct@ehbio:~/sxbd$ awk -f grade.awk de_gene- awk给每行增加行号,使其变为唯一
3.2.3 awk糅合操作 - 命令组合体现魅力
- awk中执行系统命令 (注意引号的使用)
# input_mat
ct@ehbio:~/sxbd$ cat <<END | sed 's/ */\t/g' >input_mat
SRR1 root
SRR2 leaf
SRR3 stem
END
ct@ehbio:~/sxbd$ touch SRR1.fq SRR2.fq SRR3.fq
ct@ehbio:~/sxbd$ ls
SRR1.fq SRR2.fq SRR3.fq
# 系统命令组成字符串,交给system函数运行
ct@ehbio:~/sxbd$ awk 'BEGIN{OFS=FS="\t"}{system("mv "$1".fq "$2".fq");}' input_mat
#
ct@ehbio:~/sxbd$ ls
leaf.fq root.fq stem.fq- awk 引用系统变量
3.3 SED命令 - 文本替换舍我其谁
3.3.1 sed基本参数解释
sed是stream editor的简称,擅长对文件进行各种正则操作、插入操作、替换操作和删除操作,可以全局,可以指定特定范围的行或者特定特征的行。
s/pat/replace/: 正则替换
前插行i, 后插行a, 替换行c, 删除行d, 输出行p
N: 读入下一行,同时存储;n:读入下一行,抛弃当前行
3.3.2 常见操作
- 替换特定的文本
# 空格是我们不太喜欢出现在文件中的一个符号,尤其是作为列名字时
# 列使用TAB键分割
ct@ehbio:~/sxbd$ cat <<END | sed 's/;/\t/g' mat
ID;2 cell;4 cell;8 cell;embryo
Pou5f1_1;2;3;4;5
Nanog_1;2;3.2;4.3;5
c-Myc;2;3;4;5
Tet1_3;2;3;4;5
END
# 一次替换
ct@ehbio:~/sxbd$ sed 's/ /_/' mat
ID 2_cell 4 cell 8 cell embryo
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
c-Myc 2 3 4 5
Tet1_3 2 3 4 5
# 全部替换
ct@ehbio:~/sxbd$ sed 's/ /_/g' mat
ID 2_cell 4_cell 8_cell embryo
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
c-Myc 2 3 4 5
Tet1_3 2 3 4 5- 获得逗号分隔的一组数
- 针对指定行替换
ct@ehbio:~/sxbd$ sed '2,$ s/_[0-9]//g' mat
ID 2 cell 4 cell 8 cell embryo
Pou5f1 2 3 4 5
Nanog 2 3.2 4.3 5
c-Myc 2 3 4 5
Tet1 2 3 4 5- 替换特定出现位置
# 替换第一个空格
ct@ehbio:~/sxbd$ sed 's/ /_/1' mat
ID 2_cell 4 cell 8 cell embryo
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
c-Myc 2 3 4 5
Tet1_3 2 3 4 5
# 替换第二个空格
ct@ehbio:~/sxbd$ sed 's/ /_/2' mat
ID 2 cell 4_cell 8 cell embryo
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
c-Myc 2 3 4 5
Tet1_3 2 3 4 5
# 替换第二个及以后的空格
ct@ehbio:~/sxbd$ sed 's/ /_/2g' mat
ID 2 cell 4_cell 8_cell embryo
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
c-Myc 2 3 4 5
Tet1_3 2 3 4 5- 给序列起名字
ct@ehbio:~/sxbd$ cat seq
ACDGTFGGCATGCDTGD
ACDGAGCDTAGCDGTA
CAGDTAGDCTADTG
ct@ehbio:~/sxbd$ sed = seq
1
ACDGTFGGCATGCDTGD
2
ACDGAGCDTAGCDGTA
3
CAGDTAGDCTADTG
# 同时缓冲两行,但只对第一行行首操作
ct@ehbio:~/sxbd$ sed = seq | sed 'N;s/^/>/;'
>1
ACDGTFGGCATGCDTGD
>2
ACDGAGCDTAGCDGTA
>3
CAGDTAGDCTADTG- 给文件增加标题行
ct@ehbio:~/sxbd$ tail -n +2 mat | sort -k2,2n
c-Myc 2 3 4 5
Nanog_1 2 3.2 4.3 5
Pou5f1_1 2 3 4 5
Tet1_3 2 3 4 5
# 1 表示第一行
# i 表示插入,在指定行前面插入新行
ct@ehbio:~/sxbd$ tail -n +2 mat | sort -k2,2n | sed '1 i ID\t2_cell\t4_cell\t8_cell\tembryo'
ID 2_cell 4_cell 8_cell embryo
c-Myc 2 3 4 5
Nanog_1 2 3.2 4.3 5
Pou5f1_1 2 3 4 5
Tet1_3 2 3 4 5- 提取特定或指定范围的行
# -n是必须的,阻止程序自动输出匹配行,不然会导致重复输出
ct@ehbio:~/sxbd$ sed -n '2,4p' mat
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
c-Myc 2 3 4 5
ct@ehbio:~/sxbd$ sed -n '4p' mat
c-Myc 2 3 4 5提取符合特定模式的行
/pattern/支持普通字符串和正则表达式匹配
ct@ehbio:~/sxbd$ sed -n '/_/ p' mat
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
Tet1_3 2 3 4 5
ct@ehbio:~/sxbd$ sed -n '/-/ p' mat
c-Myc 2 3 4 5- 去除文件中的空行
ct@ehbio:~/sxbd$ cat mat
ID 2 cell 4 cell 8 cell embryo
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
c-Myc 2 3 4 5
Tet1_3 2 3 4 5
# 空行就是只有行首和行尾的行
ct@ehbio:~/sxbd$ sed '/^$/d' mat
ID 2 cell 4 cell 8 cell embryo
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
c-Myc 2 3 4 5
Tet1_3 2 3 4 5- 原位删除
ct@ehbio:~/sxbd$ cat mat
ID 2 cell 4 cell 8 cell embryo
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
c-Myc 2 3 4 5
Tet1_3 2 3 4 5
# -i 参数的使用
ct@ehbio:~/sxbd$ sed -i '/^$/d' mat
ct@ehbio:~/sxbd$ cat mat
ID 2 cell 4 cell 8 cell embryo
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
c-Myc 2 3 4 5
Tet1_3 2 3 4 5- 删除指定范围的行
ct@ehbio:~/sxbd$ cat mat
ID 2 cell 4 cell 8 cell embryo
Pou5f1_1 2 3 4 5
Nanog_1 2 3.2 4.3 5
c-Myc_2 2 3 4 5
Tet1_3 2 3 4 5
ct@ehbio:~/sxbd$ sed '2,3d' mat
ID 2 cell 4 cell 8 cell embryo
c-Myc_2 2 3 4 5
Tet1_3 2 3 4 5- 记忆匹配
\(\)启动记忆匹配;\1为第一个匹配项,\2为第二个匹配项;匹配项的计数根据左括号出现的位置来定,第一个(包括起来的为\1。
- 奇偶数行处理
ct@ehbio:~/sxbd$ echo -e "odd\neven\nodd\neven"
odd
even
odd
even
# 奇偶数行合并
ct@ehbio:~/sxbd$ echo -e "odd\neven\nodd\neven" | sed 'N;s/\n/\t/'
odd even
odd even
# 取出偶数行,比较简单
# 注意 n (小写)撇掉了奇数行
ct@ehbio:~/sxbd$ echo -e "odd\neven\nodd\neven" | sed -n 'n;p'
even
even
# 取出奇数行
# 先都读进去,然后替换偶数行为空值,再输出
ct@ehbio:~/sxbd$ echo -e "odd\neven\nodd\neven" | sed -n 'N;s/\n.*//;p'
odd
odd- Windows/Linux换行符困境
Windows下的换行符是\r\n, Linux下换行符是\n, MAC下换行符是\r。所以Windows下的文件拷贝到Linux后,常会出现行尾多一个^M符号的情况,从而引起匹配或其它解析问题。
^M的输是 ctrl+v+M ctrl+v;ctrl+m,不是简单的输入^,再输入M。
ct@ehbio:~/sxbd$ cat -A windows.txt
ID^M$
A^M$
B^M$
C^M$
ct@ehbio:~/sxbd$ sed 's/^M//' windows.txt | cat -A
ID$
A$
B$
C$- sed中使用bash变量
3.4 VIM的使用
VIM是一款功能强大的文本编辑工具,也是我在Linux,Windows下编辑程序和文本最常用的工具。
3.4.1 初识VIM
VIM分多种状态模式,写入模式,正常模式,可视化模式。
正常模式:打开或新建文件默认在正常模式,可以浏览,但不可以写入内容。这个模式也可以称作命令行模式,这个模式下可以使用VIM强大的命令行和快捷键功能。其它模式下按ESC就可以到正常模式。写入模式:在正常模式下按字母i(光标前插入),o(当前光标的下一行操作),O(当前光标的上一行操作),a(光标后插入)都可以进入写入模式,就可以输入内容了。可视化模式:通常用于选择特定的内容。
进入写入模式后,VIM使用起来可以跟记事本一样了。在写入文字时,可以利用组合键CTRL+n和CTRL+p完成写作单词的自动匹配补全,从而加快输入速度,保证输入的前后一致。
正常模式有更强大的快捷键编辑功能,把手从鼠标上解放出来。
dd: 删除一行3dd: 删除一行dw: 删除一个单词d3w: 删除3个单词yy: 复制一行3yy: 复制三行yw: 复制一个单词p: (小写p)粘贴到下一行P: (大写P)粘贴到上一行>>: 当前行右缩进一个TAB3>>: 当前行及后2行都向右缩进一个TAB<<: 当前行左缩进一个TAB3<<: 当前行及后2行都向左缩进一个TAB/word: 查找特定单词u: 撤销上一次操作.: 重复上一次操作CTRL+r: 重做撤销的操作y$: 从当前复制到行尾d$: 从当前删除到行尾
跳转操作
gg: 跳到文件开头G: 跳到文件结尾zt: 当前行作为可视屏幕的第一行5G: 跳到第5行
正常模式下输入冒号进入更强大的命令行定制功能。
:5d: 删除第5行:20,24y:复制20到24行:.,+3y:复制当前行和下面3行:2,11>: 右缩进:w: 保存文件:q: 退出编辑器:vsplit: 分屏
键盘操作不容易被捕获,看右下角可以得到一点信息。动图请点击查看。

VIM还有不少魔性操作,具体可以看这两个帖子:
3.4.2 VIM中使用正则表达式
这儿以提取生信宝典公众号中发过的原创文章的HTML代码为例子,获得原创文章的名字和链接,用以制作文章列表。
部分数据如下所示,利用正则表达式的第一步就是找规律。
- 这段文字是JSON格式,列表和字典的组合,使用
json函数可以很容易解析。但我们这通过正则表达式解析。 title后面跟随的文章的题目;url后面跟随的是文章的链接。{"和"}标记每篇文章的信息的开始和结束。auth_apply_num是目前不关注的信息。
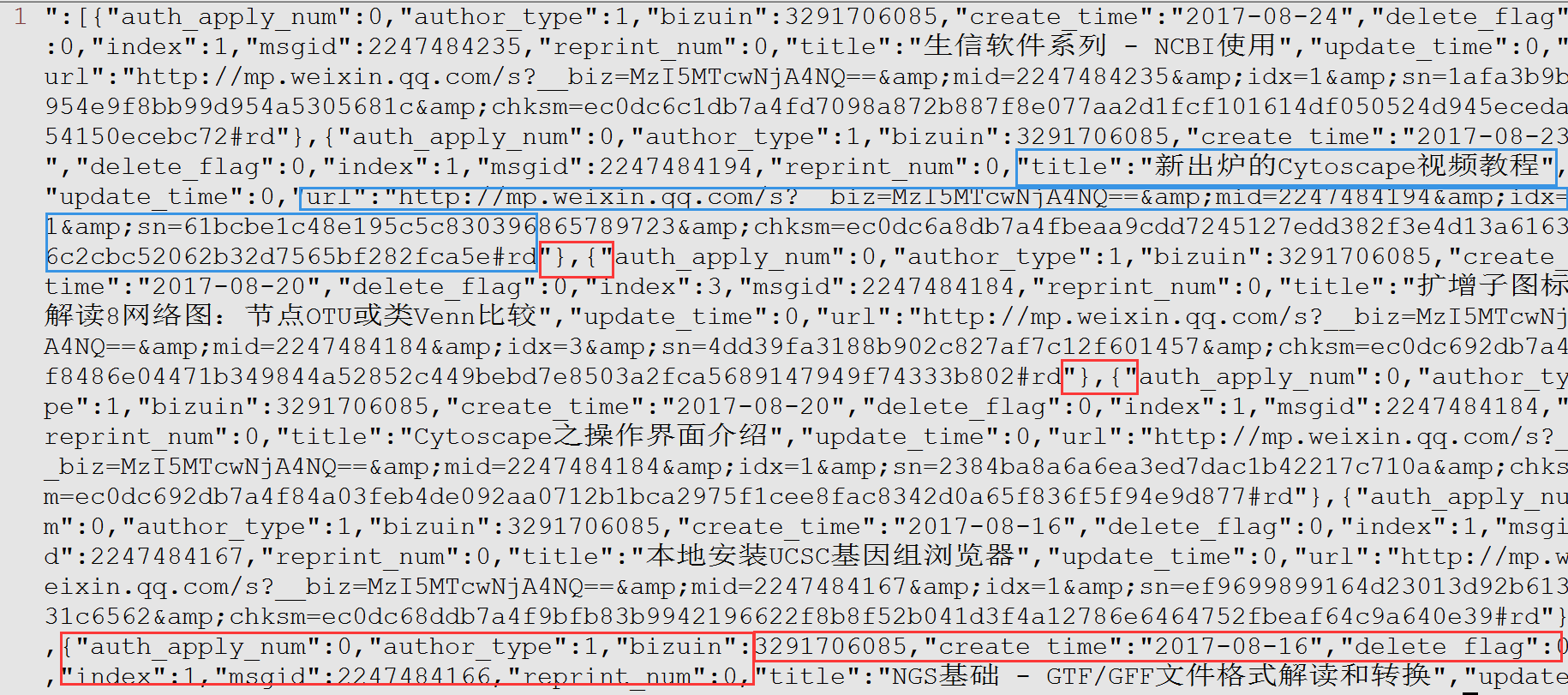
下面的动画展示了如何通过正则表达式,把这段文字只保留题目和链接,并转成Markdown的格式。
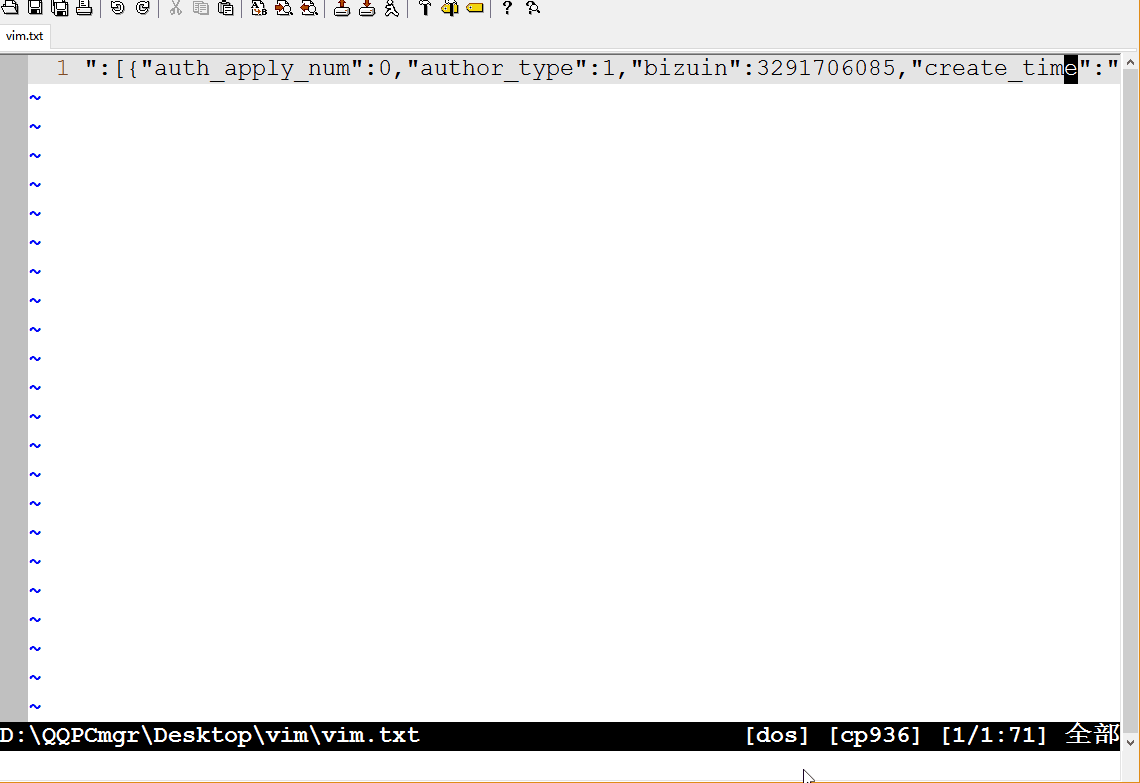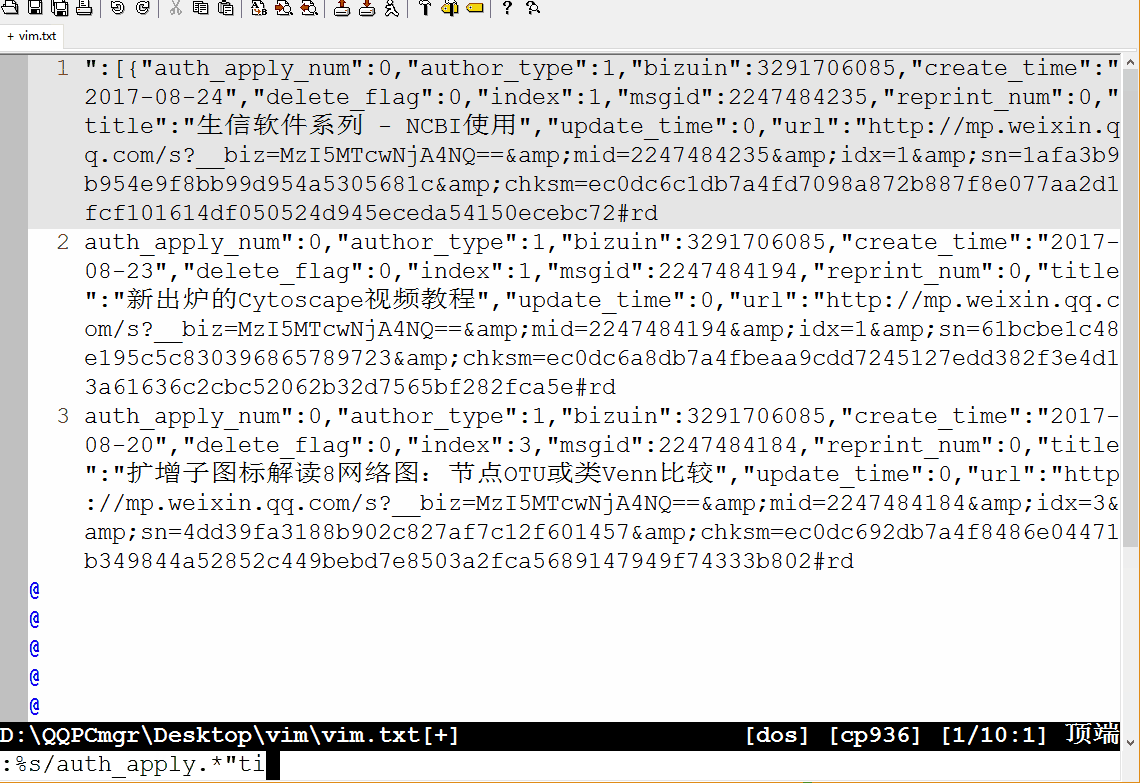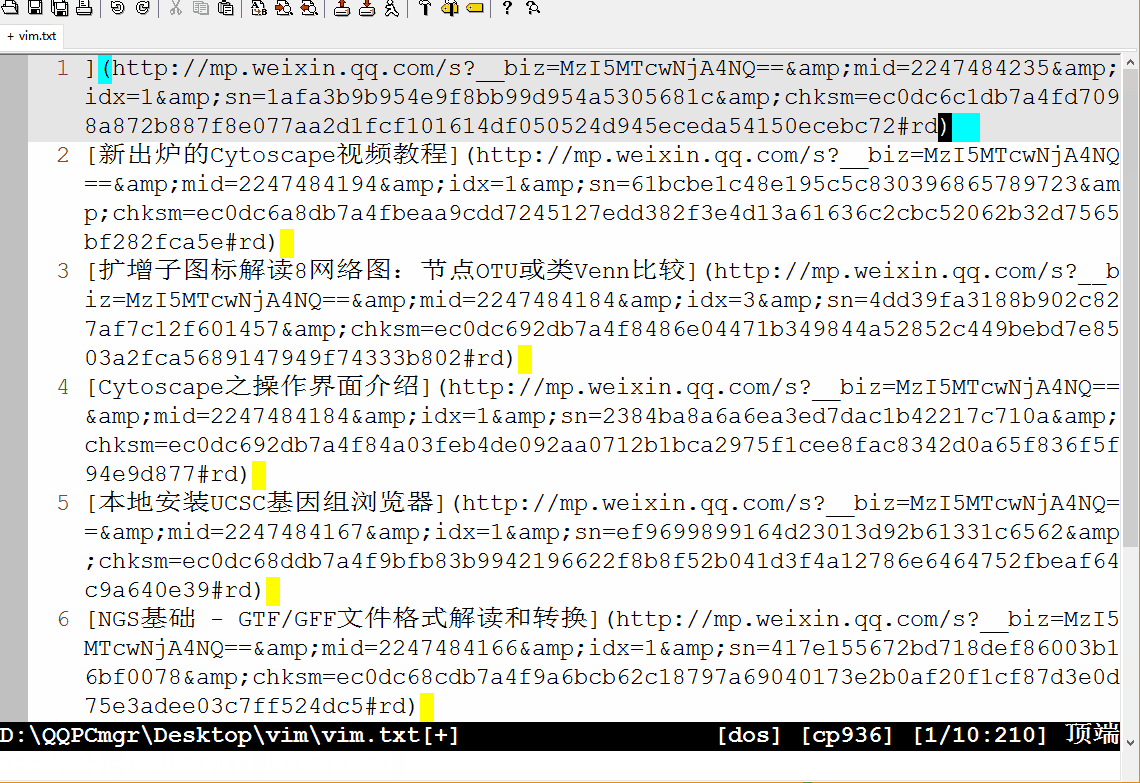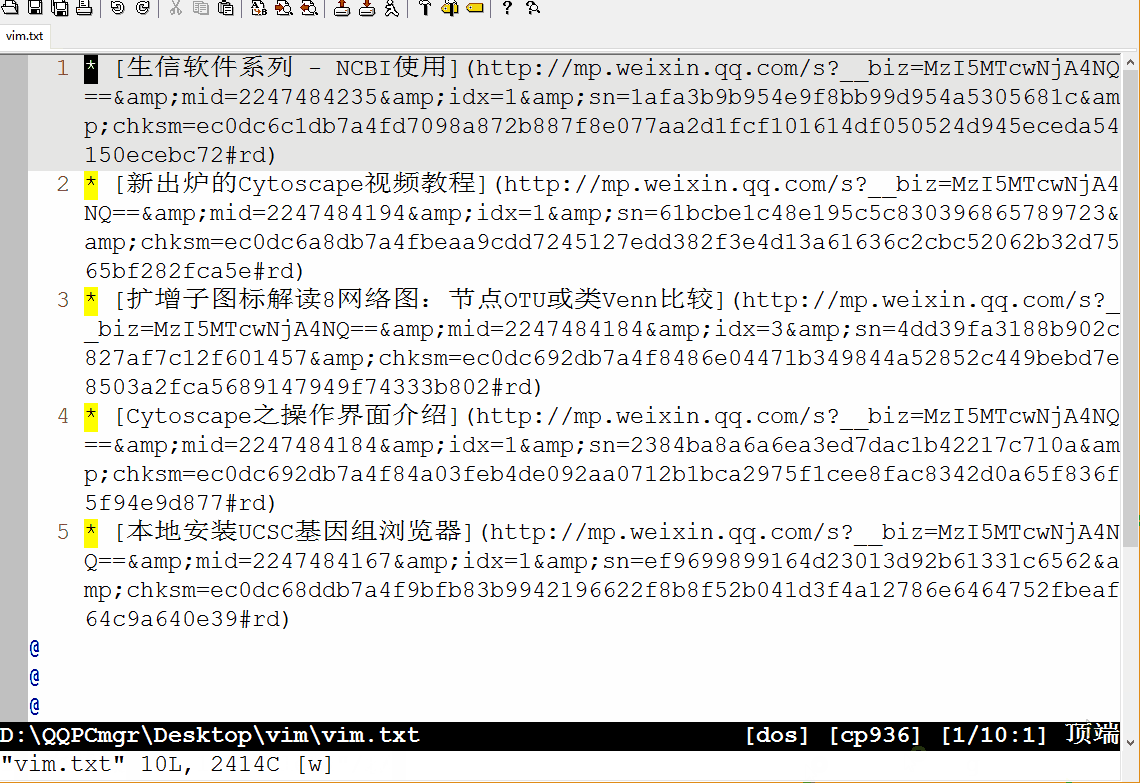
:set wrap: 折行显示:s/"}, {"/\r/g::开启命令行模式;s: 是替换,之前讲Linux命令时也多次提及;/作为分割符,三个一起出现,前两个/中的内容为被替换内容,后两个/中的内容为替换成的内容;这里没有使用正则表达式,直接是原字符的替换,\r表示换行符。这样把每篇文章的信息单行显示,方便后续处理。:%s/auth_apply.*"title":"/[/:%表示对所有行进行操作;被替换的内容是auth_apply和title":"及其之间的内容(.*表示,.表示任意字符,*表示其前面的字符出现任意次):%s/".*"url":"/](/:从题目到url之间的内容替换掉;第一次替换时忘记了第一行中开头还有引号,结果出现了误操作,后面又退回去,手动删除特殊部分,其它部分继续匹配。:%s/$/)/:表示在行尾($)加上), 就组成了Markdown中完整的链接形式[context](link)。:%s/^/* /:表示在行首(^)加上*变成Markdown格式的列表
至此就完成了生信宝典公众号文章到Markdown链接的转换,可以放到菜单栏文章集锦里面方便快速查询了。
一步步的处理也有些麻烦,有没有办法更简单些呢? 动画可查看链接。

- 首先也是把每篇文章的信息处理为单行显示,一样的模式更容易操作,去掉第一行行首不一致的部分
- 使用上下箭头可以回溯之前的命令,类似于Linux终端下的操作
%s/.*title":"\([^"]*\).*url":"\(.*\)/* [\1](\2)/c: 这个是记忆匹配,记录下匹配的内容用于替换,\(和\)表示记忆匹配的开始和结束,自身不匹配任何字符,只做标记使用;从左只右, 第一个\(中的内容记录为\1, 第二个\(中的内容记录为\2,以此类推。尤其在存在括号嵌套的情况下,注意匹配位置,左括号出现的顺序为准。在匹配文章题目时使用了[^"]*而不是.*,是考虑到正则表达式的匹配是贪婪的,会囊括更多的内容进来,就有可能出现非预期情况,所以做这么个限定,匹配所有非"内容。
正则表达式在数据分析中有很多灵活的应用,可以解决复杂的字符串抽提工作。常用的程序语言或命令如pytho, R, grep, awk, sed都支持正则表达式操作,语法也大体相似。进一步学习可参考一下链接:
3.5 有了这些,文件批量重命名还需要求助其它工具吗?
3.5.1 简单重命名
Linux下文件重命名可以通过两个命令完成,mv和rename。
mv: 直接运行可以进行单个文件的重命名,如mv old_name.txt new_name.txtrename: 默认支持单个文件或有固定规律的一组文件的批量重命名,示例如下:
3.5.1.1 rename演示
使用touch新建文件,两个样品(分别是易生信a,易生信b),各自双端测序的FASTQ文件
ysx@ehbio:~/test$ touch YSX_a_1.fq.gz YSX_a_2.fq.gz YSX_b_2.fq.gz YSX_b_1.fq.gz
ysx@ehbio:~/test$ ls
YSX_a_1.fq.gz YSX_a_2.fq.gz YSX_b_1.fq.gz YSX_b_2.fq.gz把文件名中的 易生信(YSX)改为易汉博 (ehbio)
# rename '被替换文字' '要替换成的文字' 操作对象
ysx@ehbio:~/test$ rename 'YSX' 'ehbio' *.gz
ysx@ehbio:~/test$ ls
ehbio_a_1.fq.gz ehbio_a_2.fq.gz ehbio_b_1.fq.gz ehbio_b_2.fq.gz不同操作系统,rename的使用方法略有不同。印象中:
在CentOS都是上面的语法
rename old new file_list在Ubuntu都是下面的语法
rename s/old/new/ file_list
# 在Centos下,该命令未起作用
ysx@ehbio:~/test$ rename 's/ehbio_//' *
ysx@ehbio:~/test$ ls
ehbio_a_1.fq.gz ehbio_a_2.fq.gz ehbio_b_1.fq.gz ehbio_b_2.fq.gz
# 如果写的rename命令没发挥作用,使用man rename查看写看其具体使用方法, 个人经验,无外乎上面提到的两种用法。
ysx@ehbio:~/test$ man rename
# NAME
# rename - rename files
#
# SYNOPSIS
# rename [options] expression replacement file...替换后缀
# 替换后缀
ysx@ehbio:~/test$ rename 'fq' 'fastq' *.gz
ysx@ehbio:~/test$ ls
ehbio_a_1.fastq.gz ehbio_a_2.fastq.gz ehbio_b_1.fastq.gz ehbio_b_2.fastq.gz3.5.2 复杂重命名
但有时,需要重命名的文件不像上面那样有很清晰的模式,直接可以替换,需要多几步处理获得对应关系。
3.5.2.1 假如已经有对应关系
如下name.map.txt是自己手动编写的文件,a对应Control, b对应Treatment。
ysx@ehbio:~/test$ ls
name.map.txt ehbio_a_1.fastq.gz ehbio_a_2.fastq.gz ehbio_b_1.fastq.gz ehbio_b_2.fastq.gz
ysx@ehbio:~/test$ cat name.map.txt
a Control
b Treatment3.5.2.1.1 组合文件名,使用mv重命名
首先组合出原名字和最终名字
ysx@ehbio:~/test$ awk '{print "ehbio_"$1"_1.fastq.gz", "ehbio_"$2"_1.fastq.gz", "ehbio_"$1"_2.fastq.gz", "ehbio_"$2"_2.fastq.gz"}' name.map.txt
ehbio_a_1.fastq.gz ehbio_Control_1.fastq.gz ehbio_a_2.fastq.gz ehbio_Control_2.fastq.gz
ehbio_b_1.fastq.gz ehbio_Treatment_1.fastq.gz ehbio_b_2.fastq.gz ehbio_Treatment_2.fastq.gz加上mv
ysx@ehbio:~/test$ awk '{print "mv ehbio_"$1"_1.fastq.gz ehbio_"$2"_1.fastq.gz"; print "mv ehbio_"$1"_2.fastq.gz ehbio_"$2"_2.fastq.gz";}' name.map.txt
mv ehbio_a_1.fastq.gz ehbio_Control_1.fastq.gz
mv ehbio_a_2.fastq.gz ehbio_Control_2.fastq.gz
mv ehbio_b_1.fastq.gz ehbio_Treatment_1.fastq.gz
mv ehbio_b_2.fastq.gz ehbio_Treatment_2.fastq.gz可以直接拷贝上面的输出再粘贴运行,或存储为文件运行
ysx@ehbio:~/test$ awk '{print "mv ehbio_"$1"_1.fastq.gz ehbio_"$2"_1.fastq.gz"; print "mv ehbio_"$1"_2.fastq.gz ehbio_"$2"_2.fastq.gz";}' name.map.txt >rename.sh
ysx@ehbio:~/test$ #bash rename.sh也可以把print改为system直接运行
ysx@ehbio:~/test$ ls
ehbio_a_1.fastq.gz ehbio_a_2.fastq.gz ehbio_b_1.fastq.gz ehbio_b_2.fastq.gz name.map.txt rename.sh
ysx@ehbio:~/test$ awk '{system("mv ehbio_"$1"_1.fastq.gz ehbio_"$2"_1.fastq.gz"); system("mv ehbio_"$1"_2.fastq.gz ehbio_"$2"_2.fastq.gz");}' name.map.txt
ysx@ehbio:~/test$ ls
ehbio_Control_1.fastq.gz ehbio_Control_2.fastq.gz ehbio_Treatment_1.fastq.gz ehbio_Treatment_2.fastq.gz name.map.txt rename.sh3.5.2.1.2 使用rename会不会稍微简单一点?
一定注意符号匹配和避免误匹配。
# 注意引号和空格
ysx@ehbio:~/test$ awk '{print("rename "$1" "$2" *.fastq.gz"); }' name.map.txt
rename a Control *.fastq.gz
rename b Treatment *.fastq.gz
# 上面的命令有什么问题吗?
# fastq中也存在a,是否也会被替换
# ehbio中也存在b,是否也会倍替换
ysx@ehbio:~/test$ awk '{system("rename "$1" "$2" *.fastq.gz"); }' name.map.txt
# 执行后,文件名都乱套了
ysx@ehbio:~/test$ ls
ehbio_b_1.fControlstq.gz ehbio_b_2.fControlstq.gz ehTreatmentio_Control_1.fastq.gz ehTreatmentio_Control_2.fastq.gz name.map.txt rename.sh
# 再重命名回去,再次尝试
ysx@ehbio:~/test$ rename 'Control' 'a' *
ysx@ehbio:~/test$ rename 'Treatment' 'b' *
ysx@ehbio:~/test$ ls
ehbio_a_1.fastq.gz ehbio_a_2.fastq.gz ehbio_b_1.fastq.gz ehbio_b_2.fastq.gz name.map.txt rename.sh
# 重命名两侧加下划线, 这也是我们做匹配时常需要注意的,尽量限制让匹配更准确
ysx@ehbio:~/test$ awk '{system("rename _"$1"_ _"$2"_ *.fastq.gz"); }' name.map.txt
# 打印出来看下
ysx@ehbio:~/test$ awk '{print("rename _"$1"_ _"$2"_ *.fastq.gz"); }' name.map.txt
# rename _a_ _Control_ *.fastq.gz
# rename _b_ _Treatment_ *.fastq.gz
# 这次没问题了
ysx@ehbio:~/test$ ls
ehbio_Control_1.fastq.gz ehbio_Control_2.fastq.gz ehbio_Treatment_1.fastq.gz ehbio_Treatment_2.fastq.gz name.map.txt rename.sh3.5.2.2 从原文件名获取对应关系
3.5.2.2.1 基于paste
像上面自己写好对应文件是一个方法,有时也可以从文件名推测规律,生成对应文件。
如下有一堆测序原始数据,选择A组样品来查看:
# 如下有一堆测序原始数据,选择A组样品来查看
ysx@ehbio:~/test2# ls A*
A1_FRAS192317015-1a_1.fq.gz A2_FRAS192320421-1a_1.fq.gz A3_FRAS192317017-1a_1.fq.gz
A1_FRAS192317015-1a_2.fq.gz A2_FRAS192320421-1a_2.fq.gz A3_FRAS192317017-1a_2.fq.gz中间的那一串字符FRA...-是我们不需要的。
观察规律,按下划线分割(_),获取第1,3个元素;另外习惯性给生物重复前面也加上下划线(用到了sed的记忆匹配)。
ysx@ehbio:~/test2# ls A*.gz | cut -f 1,3 -d '_' | sed 's/\([A-E]\)/\1_/'
A_1_1.fq.gz
A_1_2.fq.gz
A_2_1.fq.gz
A_2_2.fq.gz把原样品名字与新样品名字对应起来,这里用到了paste和输入重定向 (<):
ysx@ehbio:~/test2# paste <(ls A*.gz) <(ls A*.gz | cut -f 1,3 -d '_' | sed 's/\([A-E]\)/\1_/')
A1_FRAS192317015-1a_1.fq.gz A_1_1_fq.gz
A1_FRAS192317015-1a_2.fq.gz A_1_2_fq.gz
A2_FRAS192320421-1a_1.fq.gz A_2_1_fq.gz
A2_FRAS192320421-1a_2.fq.gz A_2_2_fq.gz
A3_FRAS192317017-1a_1.fq.gz A_3_1_fq.gz
A3_FRAS192317017-1a_2.fq.gz A_3_2_fq.gz使用mv直接重命名 (还可以把这个脚本保存下来,保留原始名字和新名字的对应关系,万一操作错了,在看到结果异常时也可以方便回溯)
ysx@ehbio:~/test2# paste <(ls A*.gz) <(ls A*.gz | cut -f 1,3 -d '_' | sed 's/\([A-E]\)/\1_/') | sed 's#^#/bin/mv #'
/bin/mv A1_FRAS192317015-1a_1.fq.gz A_1_1_fq.gz
/bin/mv A1_FRAS192317015-1a_2.fq.gz A_1_2_fq.gz
/bin/mv A2_FRAS192320421-1a_1.fq.gz A_2_1_fq.gz
/bin/mv A2_FRAS192320421-1a_2.fq.gz A_2_2_fq.gz
/bin/mv A3_FRAS192317017-1a_1.fq.gz A_3_1_fq.gz
/bin/mv A3_FRAS192317017-1a_2.fq.gz A_3_2_fq.gz软链接也是常用的 (但一定注意源文件使用全路径)
ysx@ehbio:~/test2# paste <(ls *.gz) <(ls *.gz | sed 's/\./_/' | cut -f 1,3,4 -d '_' | sed 's/\([A-E]\)/analysis\/\1_/') | sed 's#^#ln -s `pwd`/#'
ln -s `pwd`/A1_FRAS192317015-1a_1.fq.gz analysis/A_1_1_fq.gz
ln -s `pwd`/A1_FRAS192317015-1a_2.fq.gz analysis/A_1_2_fq.gz
ln -s `pwd`/A2_FRAS192320421-1a_1.fq.gz analysis/A_2_1_fq.gz
.
.
.
ln -s `pwd`/E15_FRAS192317028-1a_1.fq.gz analysis/E_15_1_fq.gz
ln -s `pwd`/E15_FRAS192317028-1a_2.fq.gz analysis/E_15_2_fq.gz3.5.2.2.2 基于awk {rename_awk}
转换下输入数据的格式,字符处理在awk也可以操作,但我更习惯使用命令组合,每一步都用最简单的操作,不容易出错。
ysx@ehbio:~/test2# ls A*.gz | sed -e 's/\([A-E]\)/\1_/'
A_1_FRAS192317015-1a_1.fq.gz
A_1_FRAS192317015-1a_2.fq.gz
A_2_FRAS192320421-1a_1.fq.gz
A_2_FRAS192320421-1a_2.fq.gz
A_3_FRAS192317017-1a_1.fq.gz
A_3_FRAS192317017-1a_2.fq.gz
ysx@ehbio:~/test2# ls A*.gz | sed -e 's/\([A-E]\)/\1_/' -e 's/\./_./'
A_1_FRAS192317015-1a_1_.fq.gz
A_1_FRAS192317015-1a_2_.fq.gz
A_2_FRAS192320421-1a_1_.fq.gz
A_2_FRAS192320421-1a_2_.fq.gz
A_3_FRAS192317017-1a_1_.fq.gz
A_3_FRAS192317017-1a_2_.fq.gz采用awk生成对应关系
# 生成样品重复,计数出错了,每行记了一个数,而实际两行是一个样本。
ysx@ehbio:~/test2# ls A*.gz | sed -e 's/\([A-E]\)/\1_/' -e 's/\./_./' | awk 'BEGIN{OFS=" ";FS="_"}{sum[$1]+=1; print $0, $1"_"sum[$1]"_"$4$5;}'
A_1_FRAS192317015-1a_1_.fq.gz A_1_1.fq.gz
A_1_FRAS192317015-1a_2_.fq.gz A_2_2.fq.gz
A_2_FRAS192320421-1a_1_.fq.gz A_3_1.fq.gz
A_2_FRAS192320421-1a_2_.fq.gz A_4_2.fq.gz
A_3_FRAS192317017-1a_1_.fq.gz A_5_1.fq.gz
A_3_FRAS192317017-1a_2_.fq.gz A_6_2.fq.gz# 稍微改进下
ysx@ehbio:~/test2# ls A*.gz | sed -e 's/\([A-E]\)/\1_/' -e 's/\./_./' | awk 'BEGIN{OFS=" ";FS="_"}{sum[$1]+=1; print $0, $1"_"sum[$1]"_"$4$5;}'
A_1_FRAS192317015-1a_1.fq.gz A_1_1.fq.gz
A_1_FRAS192317015-1a_2.fq.gz A_2_2.fq.gz
A_2_FRAS192320421-1a_1.fq.gz A_3_1.fq.gz
A_2_FRAS192320421-1a_2.fq.gz A_4_2.fq.gz
A_3_FRAS192317017-1a_1.fq.gz A_5_1.fq.gz
A_3_FRAS192317017-1a_2.fq.gz A_6_2.fq.gz
# 记得源文件名字的替换
ysx@ehbio:~/test2# ls A*.gz | sed -e 's/\([A-E]\)/\1_/' -e 's/\./_./' | awk 'BEGIN{OFS=" ";FS="_"}{sum[$1]+=1; print $0, $1"_"sum[$1]"_"$4$5;}' | sed -e 's/_//' -e 's/_\././' -e 's#^#ln -s `pwd`/#' |head
ln -s `pwd`/A1_FRAS192317015-1a_1.fq.gz A_1_1.fq.gz
ln -s `pwd`/A1_FRAS192317015-1a_2.fq.gz A_2_2.fq.gz好了,重命名就到这了。有了这个思路,关键是如何根据自己的文件名字特征,构造对应的匹配关系。
另外,Window下使用Git for windows应该也可以实现对应的操作。
3.6 耗时很长的程序忘加nohup就运行了怎么办?
在NGS基础:测序原始数据下载一文中提到可以使用SRA-toolkit中的命令fastq-dump从NCBI下载原始测序数据,命令如下。
nohup fastq-dump -v --split-3 --gzip SRR5908360 &
nohup fastq-dump -v --split-3 --gzip SRR5908361 &这个代码,给我们4个提示:
fastq-dump不只可以转换下载好的sra文件为fastq文件,还可以顺带下载sra文件。只需提供SRR号,就可以获得FASTQ序列。不需要先调用prefetch下载,然后再转换。其它参数解释见引用文章。- 每一行命令后面
&号表示把命令放入后台运行,当前终端可以继续输入其它命令;此处也相当于实现了一个手动并行下载多样本,配合for可以自动并行下载。 nohup表示让程序在终端因人为原因或网络原因断开后不挂断,适用于运行时间比较长的命令,一般与&连用,形式如nohup 你的命令 &(注意空格的存在)。如果程序运行输出错误信息,则会写入当前目录下nohup.out文件里面,供后续查看和调试。- 经常会有一些培训班“拿来主义”比较严重,以上推文和生信宝典的其它推文都被发现过直接用于某些培训班的教材,但从未申请过授权,也未引用过出处。更有甚者,盗版易生信早期培训教案和视频,用于自己的课程或在全网发布,希望大家多多举报。
言归正传,通常我们运行程序前,会有个预判,如前面那个例子,运行时间比较长,会使用nohup 我的命令 &的形式进行运行,从而保证程序不受网络或终端异常退出的影响。
但有时也会有误判,如没想到某个程序运行了半个小时还没结束,或数据传输时网太慢,需要传输很久,这时怎么办?中止程序,然后加上nohup再从头运行?还是有更好的办法?
下面看这个例子:马上要去吃午饭了,把文件同步到另一个服务器,饭后回来继续操作:
ysx@ehbio:~/test/Bigwig$ rsync -av * ysx@46.93.19.14:/tmp
ysx@46.93.19.14's password:
sending incremental file list
test1Y_DK10.bw输入密码后,发现同步速度太慢了,1分钟只同步了1个文件,后面还有99个文件,待会离开后,如果网断了,终端退出,程序终止怎么办?同步不能完成,饭后怎么愉快的工作?
还好我们有下面的方案,一步步跟着操作,补救一下。
第一步,按ctrl+z把程序挂起,操作后屏幕会出现如下提示([1]中的1表示命令的作业号,后面会用到):
^Z
[1]+ 已停止 rsync -av * ysx@46.93.19.14:/tmp第二步(可选),用jobs命令查看下任务状态,跟刚才的屏幕提示一致,程序被暂时终止,作业号还是1:
ysx@ehbio:~/test/Bigwig$ jobs
[1]+ 已停止 rsync -av * ysx@46.93.19.14:/tmp第三步,使用bg %1命令把作业号为1的任务放入后台,并从停止状态变为运行状态,相当于加了&后接着运行。再用jobs查看,任务状态变成了运行中,这一步很关键。如果没有运行bg %1则程序处于停止状态,一直不会运行,吃几顿饭都不会运行。
ysx@ehbio:~/test/Bigwig$ bg %1
[1]+ rsync -av * ysx@46.93.19.14:/tmp &
ysx@ehbio:~/test/Bigwig$ jobs
[1]+ 运行中 rsync -av * ysx@46.93.19.14:/tmp &第四步,运行disown -h %1,表示在终端关闭时不对作业号为1的程序发送终止信号,外部因素将不影响程序的运行。通过ps命令查看下任务进程 (可选)。
ysx@ehbio:~/test/Bigwig$ disown -h %1
ysx@ehbio:~/test/Bigwig$ ps -auwx | grep 'rsync'
ysx 18214 0.0 0.0 117844 1720 ? S 09:43 0:01 rsync -av *.bw ysx@46.93.19.14:/tmp
ysx 18215 0.1 0.0 182376 8360 ? S 09:43 0:04 ssh -l ysx 46.93.19.14 rsync --server -vlogDtpre.iLsfxC . /tmp
ysx 18340 0.0 0.0 112724 984 pts/1 S+ 10:17 0:00 grep --color=auto rsync通过以上4步就完成了对这次操作的事后补救。以后遇到同类问题,试一试这个新方案吧!
同时还有5点提示:
- 例子中使用的是
rsync同步,从节省时间来看,不是一个很好的例子。因为把命令停掉再运行一次时,已经同步完整的数据不会再同步,时间损失不会太大。这也是使用同步命令rsync相比于scp的一个好处。更多同步方式见(Linux服务器数据定期同步和备份方式。 - 例子中的
rsync或其它涉及两个服务器交互的命令,都需要我们人为输入登录密码,因此直接加nohup &运行是行不通的,无法接受密码的输入。因此通过上面这个操作先在前台启动运行、输入密码,再放入后台不挂断运行。从这个角度看,是一个不错的例子。当然解决这个问题也有其它方式,具体见ssh免密码登录远程服务器。 - 如果程序运行时,已加了
&号,放入后台了,则只需运行jobs获得作业号,再运行disown不挂断即可。 - 程序作业号不一定都是
1,如果之前就有程序在后台运行,作业号相应的会自加。后面用到作业号时也需要相应修改,不要刻板总用1。 nohup和disown都可以使程序不挂断,可以获得一样的效果,但原理不太一致。nohup可以使程序忽略挂断信号(SIGHUP)或者使程序脱离终端的控制,从而终端不能再对其发送挂断信号(SIGHUP);disown则是内生于shell,告诉shell在终止时不对对应程序发送挂断信号(SIGHUP)。
3.7 References
- www.ehbio.com/Training
- Linux学习 - 常用和不太常用的实用awk命令
- Linux-总目录
- Linux-文件和目录
- Linux-文件操作
- Linux文件内容操作
- Linux-环境变量和可执行属性
- Linux - 管道、标准输入输出
- Linux - 命令运行监测和软件安装
- Linux-常见错误和快捷操作
- Linux-文件列太多,很难识别想要的信息在哪列;别焦急,看这里。
- Linux-文件排序和FASTA文件操作
- Linux-应用Docker安装软件
- Linux服务器数据定期同步和备份方式
- VIM的强大文本处理方法
- Linux - Conda软件安装方法
- 查看服务器配置信息
- Linux - SED操作,awk的姊妹篇
- Linux - 常用和不太常用的实用awk命令
- Bash概论 - Linux系列教程补充篇
- CIRCOS圈图绘制 - circos安装
- CIRCOS圈图绘制 - 最简单绘图和解释
- CIRCOS圈图绘制 - 染色体信息展示和调整
- CIRCOS增加热图、点图、线图和区块属性
- 有了这些,文件批量重命名还需要求助其它工具吗?
- 耗时很长的程序忘加nohup就运行了怎么办?