5 Bioinfo tools
5.1 寻找Cas9的同源基因并进行进化分析
见PPT
5.2 如何获取目标基因的转录因子(上)——biomart下载基因和motif位置信息
科研过程中我们经常会使用Ensembl(http://asia.ensembl.org/index.html) 网站来获取物种的参考基因组,其中BioMart工具可以获取物种的基因注释信息,以及跨数据库的ID匹配和注释等。
在参考基因组和基因注释文件一文中有详细介绍如何在Ensembel数据库中获取参考基因组和基因注释文件。(点击蓝字即可阅读)
生信分析中,想要找到感兴趣基因的转录因子结合位点,该怎么做呢?
5.2.1 1. 文件准备
首先需要准备以下3个文件,后面两个文件可以在ensembl网站中下载:
- 感兴趣基因的名称列表(1列基因名即可)
- 基因组中各基因位置信息列表(6列的bed文件)
- 基因组中各转录因子结合位点信息列表(5列的bed文件)
5.2.2 2. 什么是bed文件?
bed格式文件提供了一种灵活的方式来定义数据行,以此描述基因注释的信息。BED行有3个必须的列和9个可选的列。 每行的数据格式要求一致。
关于bed文件格式的介绍,在https://genome.ucsc.edu/FAQ/FAQformat.html#format1中有详细说明。
我们需要下载的基因位置信息列表是一个6列的bed文件,每列信息如下:
| Chromosome/scaffold name | Gene start (bp) | Gene end (bp) | Gene stable ID | Gene name | Strand |
|---|---|---|---|---|---|
| 染色体的名称(例如chr3) | Gene起始位点 | Gene终止位点 | Gene stable ID | Gene name | 定义基因所在链的方向,+或- |
注:起始位置和终止位置以0为起点,前闭后开。
转录因子结合位点列表是一个5列的bed文件,每列信息如下:
| Chromosome/scaffold name | Start (bp) | End (bp) | Score | Feature Type |
|---|---|---|---|---|
| 染色体的名称(例如chr3) | TF起始位点 | TF终止位点 | Score | 转录因子的名字 |
具体内容见后面示例,更方便理解。
5.2.3 3. BioMart数据下载
进入Ensembl主页后点击BioMart

BioMart
使用下拉框-
CHOOSE DATASET- 选择数据库,我们选则Ensembl Genes 93;这时出现新的下拉框-CHOOSE DATASET- ,选择目的物种,以Human gene GRCh38.p12为例。如果自己实际操作,需要选择自己的数据常用的基因组版本。如果没有历史包袱,建议选择GRCh38最新版。
BioMart
选择数据库后,点击Filters对数据进行筛选,如果是对全基因组进行分析可不用筛选, 略过不填。

BioMart
点击Attributes,在GENE处依次选择1-6列的内容,勾选顺序便是结果矩阵中每列的顺序。

BioMart
如上图中所示,点击results后跳转下载页面,中间展示了部分所选的数据矩阵,确定格式无误后点击GO即可下载。

BioMart
转录因子结合位点矩阵的下载类似上面,不过在下拉框-CHOOSE DATASET- 选择数据库时,我们选则Ensembl Regulation 93,再选择Human Binding Motif (GRCh38.p12)

BioMart
在Attributes处选择需要的信息列,点击Results和GO进行数据下载

BioMart

BioMart
将上述下载的两个文件分别命名为 GRCh38.gene.bed和 GRCh38.TFmotif_binding.bed ,在Shell中查看一下:
基因组中每个基因所在的染色体、位置和链的信息,以及对应的ENSG编号和Gene symbol。
Chromosome/scaffold name Gene start (bp) Gene end (bp) Gene stable ID Gene
3 124792319 124792562 ENSG00000276626 RF00100 -1
1 92700819 92700934 ENSG00000201317 RNU4-59P -1
14 100951856 100951933 ENSG00000200823 SNORD114-2 1
22 45200954 45201019 ENSG00000221598 MIR1249 -1
1 161699506 161699607 ENSG00000199595 RF00019 1第五列为人中的转录因子,每一行表示每个转录因子在基因组范围的结合位点分布,即其可能在哪些区域有结合motif。这些区域是与TF的结合motif矩阵相似性比较高的区域,被视为潜在结合位点。有程序MEME-FIMO或Homer-Findmotifs.pl可以完成对应的工作。
Chromosome/scaffold name Start (bp) End (bp) Score Feature Type
14 23034888 23034896 7.391 THAP1
3 10026599 10026607 7.054 THAP1
10 97879355 97879363 6.962 THAP1
3 51385016 51385024 7.382 THAP1
16 20900537 20900545 6.962 THAP15.3 如何获取目标基因的转录因子(下)——Linux命令获取目标基因TF
我们知道有很多数据库可以查找启动子、UTR、TSS等区域以及预测转录因子结合位点,但是怎么用Linux命令处理基因信息文件来得到关注基因的启动子和启动子区结合的TF呢?
5.3.1 1. 基础回顾
转录起始位点(TSS):转录时,mRNA链第一个核苷酸相对应DNA链上的碱基,通常为一个嘌呤;(不考虑转录启动复合体的预转录情况)
启动子(promoter):与RNA聚合酶结合并能起始mRNA合成的序列。与传统的核心启动子概念不同,做生信分析时,一般选择转录起始位点上游1 kb,下游 200 nt,也有选上下游各1 kb或者 2 kb的(记住这两个数,之后计算要用到);总体上生信中选择的启动子更长,范围更广一些。
文件准备:感兴趣的基因列表(命名为targetGene.list)、还有上一期下载的GRCh38.gene.bed和GRCh38.TFmotif_binding.bed
5.3.2 2. 文件格式处理
删除文件GRCh38.gene.bed首行,第六列正负链表示形式改为-和+,并在第一列染色体位置加上chr;
sed -i '1d' GRCh38.gene.bed
# 如果用sed,注意下面2列的顺序,为什么不能颠倒过来?
sed -i 's/-1$/-/' GRCh38.gene.bed
sed -i 's/1$/+/' GRCh38.gene.bed
sed -i 's/^/chr/' GRCh38.gene.bed删除文件GRCh38.TFmotif_binding.bed首行,并在第一列染色体位置加上chr
sed -i '1d' GRCh38.TFmotif_binding.bed
sed -i 's/^/chr/' GRCh38.TFmotif_binding.bedless -S filename查看一下两个矩阵内容,发现已转换完成
chr3 124792319 124792562 ENSG00000276626 RF00100 -
chr1 92700819 92700934 ENSG00000201317 RNU4-59P -
chr14 100951856 100951933 ENSG00000200823 SNORD114-2 +
chr22 45200954 45201019 ENSG00000221598 MIR1249 -
chr1 161699506 161699607 ENSG00000199595 RF00019 +chr14 23034888 23034896 7.391 THAP1
chr3 10026599 10026607 7.054 THAP1
chr10 97879355 97879363 6.962 THAP1
chr3 51385016 51385024 7.382 THAP1
chr16 20900537 20900545 6.962 THAP15.3.3 3. 计算基因的启动子区
上面已提过,根据经验一般启动子区域在转录起始位点(TSS)上游1 kb、下游 200 nt处,注意正负链的运算方式是不一样的,切忌出错。
awk 'BEGIN{OFS=FS="\t"}{if($6=="+") {tss=$2; tss_up=tss-1000; tss_dw=tss+200;} else {tss=$3; tss_up=tss-200; tss_dw=tss+1000;} if(tss_up<0) tss_up=0;print $1, tss_up, tss_dw,$4,$5,$6;}' GRCh38.gene.bed > GRCh38.gene.promoter.U1000D200.bed关于awk命令的使用方法,可以参考Linux学习 - 常用和不太常用的实用awk命令一文。
head GRCh38.gene.bed GRCh38.gene.promoter.U1000D200.bed检查一下计算是否有误。自己选取正链和负链的一个或多个基因做下计算,看看结果是否一致。做分析不是出来结果就完事了,一定谨防程序中因为不注意核查引起的bug。
==> GRCh38.gene.bed <==
chr3 124792319 124792562 ENSG00000276626 RF00100 -
chr1 92700819 92700934 ENSG00000201317 RNU4-59P -
chr14 100951856 100951933 ENSG00000200823 SNORD114-2 +
chr22 45200954 45201019 ENSG00000221598 MIR1249 -
chr1 161699506 161699607 ENSG00000199595 RF00019 +
==> GRCh38.gene.promoter.U1000D200.bed <==
chr3 124792362 124793562 ENSG00000276626 RF00100 -
chr1 92700734 92701934 ENSG00000201317 RNU4-59P -
chr14 100950856 100952056 ENSG00000200823 SNORD114-2 +
chr22 45200819 45202019 ENSG00000221598 MIR1249 -
chr1 161698506 161699706 ENSG00000199595 RF00019 +5.3.4 4. 取两文件的交集
本条命令我们使用了bedtools程序中的子命令intersect
intersect可用来求区域之间的交集,可以用来注释peak,计算reads比对到的基因组区域不同样品的peak之间的peak重叠情况;Bedtools使用简介一文中有关于bedtools的详细介绍;
两文件取完交集后,cut -f取出交集文件的第5列和第11列,sort -u去处重复项,并将这两列内容小写全转变为大写,最终得到一个两列的文件。第一列是基因名,第二列是能与基因结合的TF名字。
程序不细解释,具体看文后的Linux系列教程。Bedtools使用简介
# cut时注意根据自己的文件选择对应的列
# tr转换大小写。
bedtools intersect -a GRCh38.gene.promoter.U1000D200.bed -b GRCh38.TFmotif_binding.bed -wa -wb | cut -f 5,11 | sort -u | tr 'a-z' 'A-Z' > GRCh38.gene.promoter.U1000D200.TF_binding.txt5.3.5 5. 提取我们关注的基因
上一步中,我们将GRCh38.gene.promoter.U1000D200.TF_binding.txt文件中的基因名和TF名都转换成了大写。
为了接下来提取目标基因转录因子时不会因大小写差别而漏掉某些基因,我们将targetGene.list中的基因名也全部转换成大写。
# 基因名字转换为大写,方便比较。不同的数据库不同的写法,只有统一了才不会出现不必要的失误
tr 'a-z' 'A-Z' targetGene.list > GeneUP.list目标基因列表和基因-TF对应表都好了,内容依次如下:
==> GeneUP.list <==
ACAT2
ACTA1
ACTA2
ADM
AEBP1
==> GRCh38.gene.promoter.U1000D200.TF_binding.txt <==
A1BG RXRA
A2M-AS1 GABP
A2M SRF
A4GALT GABP
AAAS CTCF用awk命令,根据第一个文件GeneUP.list建立索引,若第二个文件GRCh38.gene.promoter.U1000D200.TF_binding.txt第一列中检索到第一个文件中的基因,则把第二个文件中检索到目标基因的整行存储起来,最终得到了目标基因和基因对应TF的文件targetGene.TF_binding.txt。这也是常用的取子集操作。
awk 'BEGIN{OFS=FS="\t"}ARGIND==1{save[$1]=1;}ARGIND==2{if(save[$1==1]) print $0}' GeneUP.list GRCh38.gene.promoter.U1000D200.TF_binding.txt > targetGene.TF_binding.txt获取目标基因的转录因子是生信分析中常见的分析,希望如何获取目标基因的转录因子(上)和本文能够帮助到各位小伙伴
5.3.6 重点总结
什么是bed文件
awk命令的使用
bedtools使用 (Bedtools使用简介)
5.4 emboss的使用
EMBOSS是欧洲分子生物学开放软件包,主要做序列比对,数据库搜搜,蛋白motif分析和功能域分析,序列模式搜索,引物设计等。
| Popular.applications | Functions |
|---|---|
| prophet | Gapped alignment for profiles. |
| infoseq | Displays some simple information about sequences. |
| water | Smith-Waterman local alignment. |
| pepstats | Protein statistics. |
| showfeat | Show features of a sequence. |
| palindrome | Looks for inverted repeats in a nucleotide sequence. |
| eprimer3 | Picks PCR primers and hybridization oligos. |
| profit | Scan a sequence or database with a matrix or profile. |
| extractseq | Extract regions from a sequence. |
| marscan | Finds MAR/SAR sites in nucleic sequences. |
| tfscan | Scans DNA sequences for transcription factors. |
| patmatmotifs | Compares a protein sequence to the PROSITE motif database. |
| showdb | Displays information on the currently available databases. |
| wossname | Finds programs by keywords in their one-line documentation. |
| abiview | Reads ABI file and display the trace. |
| tranalign | Align nucleic coding regions given the aligned proteins. |
emboss可以使用源码编译安装或用Conda安装,在前面的基础课中已有过讲述。
下载地址 ftp://emboss.open-bio.org/pub/EMBOSS/emboss-latest.tar.gz。
下载地址 http://primer3.sourceforge.net/。
# Make sure bioconda channel has added
# http://blog.genesino.com/2017/09/bioconda/
ct@ehbio:~$ conda install emboss
ct@ehbio:~$ url=https://sourceforge.net/projects/primer3/files/primer3/2.3.7/
ct@ehbio:~$ wget ${url}primer3-2.3.7.tar.gz -O primer3-2.3.7.tar.gz
ct@ehbio:~$ tar xvzf primer3-2.3.7.tar.gz
ct@ehbio:~$ cd primer3-2.3.7/src
ct@ehbio:~$ make all
# 确保~/bin在环境变量中
ct@ehbio:~$ ln -s `pwd`/primer3_core ~/bin/primer32_core
# Error: thermodynamic approach chosen, but path to thermodynamic parameters not specified
ct@ehbio:~$ mkdir /opt/primer3_config
ct@ehbio:~$ cp -R primer3-2.3.7/src/primer3_config/* /opt/primer3_config测试数据
ct@ehbio:~$ cat <<END >test.fa
>comp24_c0_seq1
TTACTCTCATCCTCCCCTTGTTGAAAGATTGGCTGCAATTGATGAACCCGATAAGAAGGTCAACTAAGAGAAGTGTAC
TTTTACGCATGGCATGGCATGGCGAGATATGGCTGTAATATGAGTATTATTTTCCTATGTTGCTACCGATATTTTCTA
TTTGCATATGAAAATTCCAAACCCAGAGTTAGGGGCCATATCTAAAGGGAATTTGCTAACGAGTAAATGGGAAAATAG
GAAATGTCAGAGGAGAtagcctagcctagcctagcctagccTCGCCTCATGTAACGAAATACAATTTAAATTTTGCTT
TACAGCTAATAGTCAGACTTTACATTTTGCTAAAA
END设计引物
ct@ehbio:~$ eprimer32 -sequence test.fa -outfile test.fa.primer \
-targetregion 0,371 -optsize 20 -numreturn 3 \
-minsize 15 -maxsize 25 \
-opttm 50 -mintm 45 -maxtm 55 \
-psizeopt 200 -prange 100-280引物结果
# EPRIMER32 RESULTS FOR comp24_c0_seq1
# Start Len Tm GC% Sequence
1 PRODUCT SIZE: 200
FORWARD PRIMER 126 20 50.17 35.00 TATTTTCCTATGTTGCTACC
REVERSE PRIMER 306 20 50.01 30.00 ACTATTAGCTGTAAAGCAAA
2 PRODUCT SIZE: 199
FORWARD PRIMER 134 20 49.88 30.00 TATGTTGCTACCGATATTTT
REVERSE PRIMER 313 20 50.30 35.00 AAGTCTGACTATTAGCTGTA
3 PRODUCT SIZE: 198
FORWARD PRIMER 134 20 49.88 30.00 TATGTTGCTACCGATATTTT
REVERSE PRIMER 312 20 50.30 35.00 AGTCTGACTATTAGCTGTAA整理引物格式位PrimerSearch需要的格式
ct@ehbio:~$ awk '{if($0~/EPRIMER32/) {seq_name=$5;count=1;} else \
if($0~/FORWARD PRIMER/) forward=$7; else if ($0~/REVERSE PRIMER/) \
{reverse=$7; printf("%s@%d\t%s\t%s\n", seq_name,count,forward, reverse); \
count+=1;} }' test.fa.primer >all_primer_filect@ehbio:~$ cat all_primer_file
comp24_c0_seq1@1 TATTTTCCTATGTTGCTACC ACTATTAGCTGTAAAGCAAA
comp24_c0_seq1@2 TATGTTGCTACCGATATTTT AAGTCTGACTATTAGCTGTA
comp24_c0_seq1@3 TATGTTGCTACCGATATTTT AGTCTGACTATTAGCTGTAA模拟PCR
ct@ehbio:~$ primersearch -seqall test.fa -infile all_primer_file \
-mismatchpercent 5 -outfile test.database.primerSearchPrimer name comp24_c0_seq1@1
Amplimer 1
Sequence: comp24_c0_seq1
TATTTTCCTATGTTGCTACC hits forward strand at 126 with 0 mismatches
ACTATTAGCTGTAAAGCAAA hits reverse strand at [23] with 0 mismatches
Amplimer length: 200 bp
Primer name comp24_c0_seq1@2
Amplimer 1
Sequence: comp24_c0_seq1
TATGTTGCTACCGATATTTT hits forward strand at 134 with 0 mismatches
AAGTCTGACTATTAGCTGTA hits reverse strand at [16] with 0 mismatches
Amplimer length: 199 bp
Primer name comp24_c0_seq1@3
Amplimer 1
Sequence: comp24_c0_seq1
TATGTTGCTACCGATATTTT hits forward strand at 134 with 0 mismatches
AGTCTGACTATTAGCTGTAA hits reverse strand at [17] with 0 mismatches
Amplimer length: 198 bpneedleall 读入两个文件,第一个文件的每个序列都与第二个文件的每个序列进行全局比对,采用Needleman-Wunsch算法。
# 生成测试数据
ct@ehbio:~$ cat <<END >generateRandom.awk
BEGIN{srand(seed); seq[0]="A"; seq[1]="C"; seq[2]="G"; seq[3]="T"}
{for(i=1;i<=chrNum;i++)
{print ">"label""i; len=(10-int(rand()*10)%2)/10*expected_len;
for(j=0;j<=len;j++) printf("%s", seq[int(rand()*10)%4]); print "";
}
}
END
ct@ehbio:~$ echo 1 | awk -v seed=$RANDOM -v label=mm -v chrNum=2 \
-v expected_len=40 -f generateRandom.awk >test1.fa
ct@ehbio:~$ echo 1 | awk -v seed=$RANDOM -v label=hs -v chrNum=2 \
-v expected_len=40 -f generateRandom.awk >test2.fact@ehbio:~$ cat test1.fa
>mm1
GTATACATCCGTAATCGGATAAAAGCGTACTATGGCG
>mm2
TAATTTCCCATGCACTATCACAACCCCTCGGATCAGACGCC
ct@ehbio:~$ cat test2.fa
>hs1
GCAAACGATTGGCCGGACGTCATCACTCCCCTCCGCGGATG
>hs2
CACAGTCCACGCTTTAAACGTACGAACAGACTTCCTT# 输出格式见: http://emboss.sourceforge.net/docs/themes/AlignFormats.html
# Both fa and fq are supported
# -auto: 关闭弹出选项
ct@ehbio:~$ needleall -asequence test1.fa -bsequence test2.fa -gapopen 10 -gapextend 0.5 \
-outfile test12.needle.alignment -auto -aformat3 pairct@ehbio:~$ cat test12.needle.alignment
########################################
# Program: needleall
# Rundate: Fri 30 Mar 2018 13:49:30
# Commandline: needleall
# -asequence test1.fa
# -bsequence test2.fa
# -auto
# -aformat3 pair
# Align_format: pair
# Report_file: test1.needleall
########################################
#=======================================
#
# Aligned_sequences: 2
# 1: mm1
# 2: hs1
# Matrix: EDNAFULL
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 62
# Identity: 15/62 (24.2%)
# Similarity: 15/62 (24.2%)
# Gaps: 46/62 (74.2%)
# Score: 27.0
#
#
#=======================================
mm1 1 ------------------GT-ATACA------TCCGTAATCGGATAAAAG 25
|| || || |||| |||||.
hs1 1 GCAAACGATTGGCCGGACGTCAT-CACTCCCCTCCG----CGGATG---- 41
mm1 26 CGTACTATGGCG 37
hs1 42 ------------ 41
#=======================================
#
# Aligned_sequences: 2
# 1: mm2
# 2: hs1
# Matrix: EDNAFULL
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 51
# Identity: 23/51 (45.1%)
# Similarity: 23/51 (45.1%)
# Gaps: 20/51 (39.2%)
# Score: 41.0
#
#
#=======================================
mm2 1 -----TAATTTCCCATGCAC--TATCACAACCCCT---CGGATCAGACGC 40
..|||..|| |.|| .||||| .||||| |||||.
hs1 1 GCAAACGATTGGCC--GGACGTCATCAC-TCCCCTCCGCGGATG------ 41
mm2 41 C 41
hs1 42 - 41
#=======================================
#
# Aligned_sequences: 2
# 1: mm1
# 2: hs2
# Matrix: EDNAFULL
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 51
# Identity: 18/51 (35.3%)
# Similarity: 18/51 (35.3%)
# Gaps: 28/51 (54.9%)
# Score: 26.0
#
#
#=======================================
mm1 1 GTATACA-TCCGTAATCGGATAAAAGCGTACTATGGCG------------ 37
.||| ||| .||..|.||| |||| ||
hs2 1 ---CACAGTCC----ACGCTTTAAA-CGTA------CGAACAGACTTCCT 36
mm1 38 - 37
hs2 37 T 37
#=======================================
#
# Aligned_sequences: 2
# 1: mm2
# 2: hs2
# Matrix: EDNAFULL
# Gap_penalty: 10.0
# Extend_penalty: 0.5
#
# Length: 55
# Identity: 18/55 (32.7%)
# Similarity: 18/55 (32.7%)
# Gaps: 32/55 (58.2%)
# Score: 36.0
#
#
#=======================================
mm2 1 TAATTTCCCATGCACTATCACAACCC---CT-----CG---GATCAGACG 39
||||..|| || || ||.|||||.
hs2 1 ------------------CACAGTCCACGCTTTAAACGTACGAACAGACT 32
mm2 40 CC--- 41
.|
hs2 33 TCCTT 37
#---------------------------------------
#---------------------------------------ct@ehbio:~$ needleall -asequence test1.fa -bsequence test2.fa -gapopen 10 -gapextend 0.5 \
-outfile test12.needle.score -auto# 序列1 序列2 比对长度 比对得分
ct@ehbio:~$ cat test12.needle.score
mm1 hs1 62 (27.0)
mm2 hs1 51 (41.0)
mm1 hs2 51 (26.0)
mm2 hs2 55 (36.0)5.5 使用samtools计算SNP
- 安装samtools和bedtools
ct@ehbio:~$ conda install samtools
ct@ehbio:~$ conda install bedtools- 产生随机的基因组文件。
# srand: 随机数发生器。设置固定的种子,保证每次出来的结果一致
# rand: 返回[0,1)之间的随机数,包含0不包含1
ct@ehbio:~$ echo 1 | awk -v seed=1 -v label=chr -v chrNum=4 \
-v expected_len=60000 -f generateRandom.awk >genome.fa
# 显示前60个碱基
ct@ehbio:~$ ct@ehbio:~/bio$ head genome.fa | cut -c 1-60
>chr1
GACCCACACTACGAGGCTCCCAACGATCAGGATTCCTATTCCCTCCTCGCTACCGGAAAA
>chr2
AGCCCTTACACCATCTGAGTCTGGCACACTTTTAGAACATCTACCCGTCACGAACAAGAA
>chr3
GTACAAGGCCCGGGGCTCGGACATTAAGCTCCTCCACTCAGCAGTCAAGTCAAACGAACA
>chr4
ACGCCCGTCAATTAGAGGCATTCAAAGACACCCGCCCGTGCTACAATAGGTACTACAACC- 产生随机的测序文件
# -N: 获得40K read pairs
# mut.txt: 突变位点或区域
ct@ehbio:~$ wgsim -N 40000 genome.fa ehbio_1.fq ehbio_2.fq > mut.txt# FASTQ格式序列,4行一组
# 第一行以@开头,后面为序列名字,
# 第二行为序列
# 第三行+开头,后面一般无内容;若有,也是序列名字
# 第四行,质量值,对应序列中每个碱基的测序准确度
ct@ehbio:~$ head ehbio_1.fq
@chr1_17674_18124_2:0:0_2:0:0_0/1
TCGTTCAGTGGTGGTTACTCGTAGGGTCTTCCATCTGAGGCGGGCGAGCGGACGCCTTTTCTGCCTCCAG
+
2222222222222222222222222222222222222222222222222222222222222222222222
@chr1_29806_30221_2:0:0_0:0:0_1/1
TTAAGTGTGCTTGGACAACGGATATGCAAGTGTCTTTGATATATCGTTAGGGATAGGTTAATTAAGGGTC
+
2222222222222222222222222222222222222222222222222222222222222222222222# 获得的突变文件如下
# Check IUPAC here: http://www.bioinformatics.org/sms/iupac.html
Col1: chromosome
Col2: position
Col3: original base
Col4: new base (IUPAC codes indicate heterozygous)
Col5: which genomic copy/haplotype
ct@ehbio:~$ head mut.txt
chr1 6274 T C -
chr1 6923 C Y +
chr1 7022 C Y +
chr1 10426 A W +
chr1 11130 C S +
chr1 12135 G R +- 创建基因组索引
ct@ehbio:~$ bwa index genome.fa
# samtools fadix快速获取某区域序列
ct@ehbio:~$ samtools faidx genome.fa- 序列比对回基因组
ct@ehbio:~$ bwa mem -t 3 genome.fa ehbio_1.fq ehbio_2.fq | gzip >map.sam.gz- 筛选比对上的高质量reads
ct@ehbio:~$ samtools view -F4 -q1 -b map.sam.gz -o map.bam
# 下面2个排序用法都可以,看使用的samtools版本
ct@ehbio:~$ #samtools sort -@ 2 map.bam map.sortP
ct@ehbio:~$ samtools sort -@ 2 -o map.sortP.bam map.bam
ct@ehbio:~$ samtools index map.sortP.bam- 统计比对reads数
ct@ehbio:~$ samtools view -c map.sortP.bam
79998- 统计未比对上的reads数
ct@ehbio:~$ samtools view -c -f 4 map.sam.gz- 统计比对到正链的reads数
ct@ehbio:~$ samtools view -c -F 16 map.sam.gz
40001- 获取properly-paired的reads数
ct@ehbio:~$ samtools view -f2 -F 256 -c map.sortP.bam
79996- 查看每个位置碱基比对或错配情况
# -Q 0: 测试数据使用,默认为-Q 13,表示过滤掉低质量测序碱基
ct@ehbio:~$ samtools mpileup -f genome.fa -Q 0 map.sortP.bam | less
# chrName coordinate ref_base coverage reads_base reads_quality
chr1 5 C 1 ^]. 2
chr1 6 A 2 .^]. 22
chr1 7 C 2 .. 22
chr1 8 A 2 .. 22
chr1 9 C 2 G. 22
chr1 10 T 3 ..^]. 222
chr1 11 A 3 ... 222
chr1 12 C 4 ...^]. 2222
chr1 13 G 4 .... 2222
chr1 14 A 4 .... 2222
chr1 15 G 4 .... 2222
chr1 16 G 4 .... 2222mpileup format (http://samtools.sourceforge.net/pileup.shtml)
测序碱基列解释:
点(
.)代表匹配正链碱基逗号(
,)代表匹配负链碱基大写字母(
ACGTN)表示正链错配小写字母(
acgtn)表示负链错配模式
\+[0-9]+[ACGTNacgtn]+表示在当前参考位置和下一个参考位置之间有插入,插入碱基数是+后面的证书,插入碱基是数字后面的字母串。下面展示的是2 bp的插入seq2 156 A 11 .$……+2AG.+2AG.+2AGGG <975;:<<<<<
模式
-[0-9]+[ACGTNacgtn]+'参考基因组存在碱基缺失。下面展示的是4 bp`缺失:seq3 200 A 20 ,,,,,..,.-4CACC.-4CACC….,.,,.^~. ==<<<<<<<<<<<::<;2<<
符号
^表示测序序列起始位置落于此 (A symbol^' marks the start of a read segment which is a contiguous subsequence on the read separated byN/S/H’ CIGAR operations). 后面跟随的符号的ASCII值减去33表示该位置碱基的质量。符号`$’表示测序序列片段的终止。主要用于从pileup文件中获得原始测序reads。
- 输出vcf格式
# 获得未压缩的vcf格式,方便查看
ct@ehbio:~$ samtools mpileup -f genome.fa -Q 0 -vu map.sortP.bam >snp.vcf5.6 Bedtools使用
Bedtools是处理基因组信息分析的强大工具集合。
bedtools: flexible tools for genome arithmetic and DNA sequence analysis.
usage: bedtools <subcommand> [options]
The bedtools sub-commands include:
[ Genome arithmetic ]
intersect Find overlapping intervals in various ways.
求区域之间的交集,可以用来注释peak,计算reads比对到的基因组区域
不同样品的peak之间的peak重叠情况。
window Find overlapping intervals within a window around an interval.
closest Find the closest, potentially non-overlapping interval.
寻找最近但可能不重叠的区域
coverage Compute the coverage over defined intervals.
计算区域覆盖度
map Apply a function to a column for each overlapping interval.
genomecov Compute the coverage over an entire genome.
merge Combine overlapping/nearby intervals into a single interval.
合并重叠或相接的区域
cluster Cluster (but don't merge) overlapping/nearby intervals.
complement Extract intervals _not_ represented by an interval file.
获得互补区域
subtract Remove intervals based on overlaps b/w two files.
计算区域差集
slop Adjust the size of intervals.
调整区域大小,如获得转录起始位点上下游3 K的区域
flank Create new intervals from the flanks of existing intervals.
sort Order the intervals in a file.
排序,部分命令需要排序过的bed文件
random Generate random intervals in a genome.
获得随机区域,作为背景集
shuffle Randomly redistrubute intervals in a genome.
根据给定的bed文件获得随机区域,作为背景集
sample Sample random records from file using reservoir sampling.
spacing Report the gap lengths between intervals in a file.
annotate Annotate coverage of features from multiple files.
[ Multi-way file comparisons ]
multiinter Identifies common intervals among multiple interval files.
unionbedg Combines coverage intervals from multiple BEDGRAPH files.
[ Paired-end manipulation ]
pairtobed Find pairs that overlap intervals in various ways.
pairtopair Find pairs that overlap other pairs in various ways.
[ Format conversion ]
bamtobed Convert BAM alignments to BED (& other) formats.
bedtobam Convert intervals to BAM records.
bamtofastq Convert BAM records to FASTQ records.
bedpetobam Convert BEDPE intervals to BAM records.
bed12tobed6 Breaks BED12 intervals into discrete BED6 intervals.
[ Fasta manipulation ]
getfasta Use intervals to extract sequences from a FASTA file.
提取给定位置的FASTA序列
maskfasta Use intervals to mask sequences from a FASTA file.
nuc Profile the nucleotide content of intervals in a FASTA file.
[ BAM focused tools ]
multicov Counts coverage from multiple BAMs at specific intervals.
tag Tag BAM alignments based on overlaps with interval files.
[ Statistical relationships ]
jaccard Calculate the Jaccard statistic b/w two sets of intervals.
计算数据集相似性
reldist Calculate the distribution of relative distances b/w two files.
fisher Calculate Fisher statistic b/w two feature files.
[ Miscellaneous tools ]
overlap Computes the amount of overlap from two intervals.
igv Create an IGV snapshot batch script.
用于生成一个脚本,批量捕获IGV截图
links Create a HTML page of links to UCSC locations.
makewindows Make interval "windows" across a genome.
把给定区域划分成指定大小和间隔的小区间 (bin)
groupby Group by common cols. & summarize oth. cols. (~ SQL "groupBy")
分组结算,不只可以用于bed文件。
expand Replicate lines based on lists of values in columns.
split Split a file into multiple files with equal records or base pairs.
[ General help ]
--help Print this help menu.
--version What version of bedtools are you using?.
--contact Feature requests, bugs, mailing lists, etc.- 安装bedtools
ct@ehbio:~$ conda install bedtoolsct@ehbio:~$ mkdir bedtools
ct@ehbio:~$ cd bedtools
ct@ehbio:~$ url=https://s3.amazonaws.com/bedtools-tutorials/web
ct@ehbio:~/bedtools$ curl -O ${url}/maurano.dnaseI.tgz
ct@ehbio:~/bedtools$ curl -O ${url}/cpg.bed
ct@ehbio:~/bedtools$ curl -O ${url}/exons.bed
ct@ehbio:~/bedtools$ curl -O ${url}/gwas.bed
ct@ehbio:~/bedtools$ curl -O ${url}/genome.txt
ct@ehbio:~/bedtools$ curl -O ${url}/hesc.chromHmm.bed- 交集 (intersect)
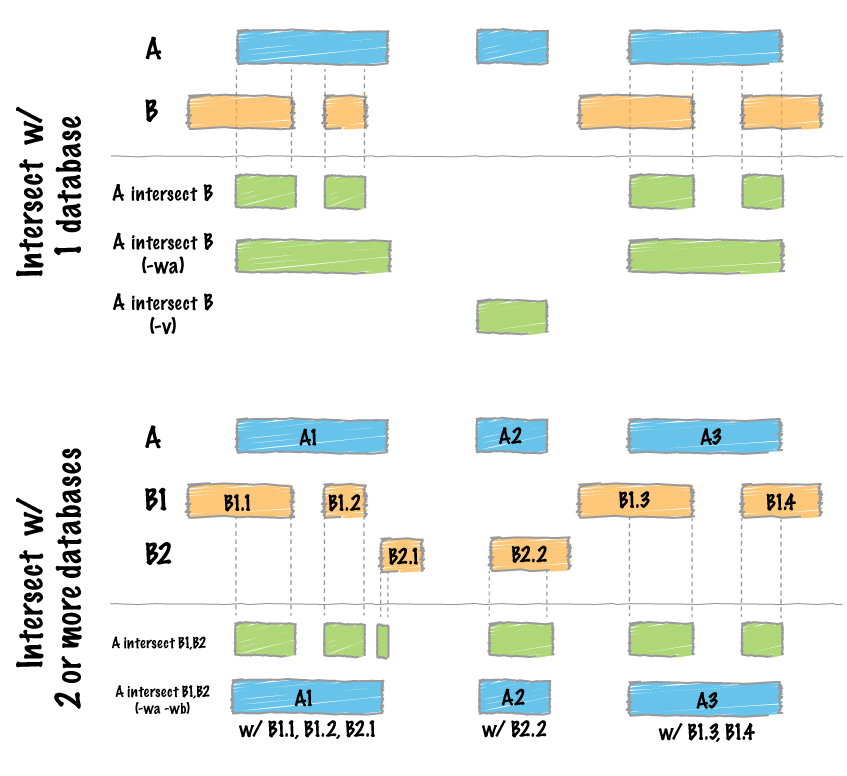
Figure 5.1: bedtools intersect
查看输入文件,bed格式,至少三列,分别是染色体,起始位置(0-based, 包括),终止位置 (1-based,不包括)。第四列一般为区域名字,第五列一般为空,第六列为链的信息。更详细解释见http://www.genome.ucsc.edu/FAQ/FAQformat.html#format1。
自己做研究CpG岛信息可以从UCSC的Table Browser获得,具体操作见http://blog.genesino.com/2013/05/ucsc-usages/。
ct@ehbio:~/bedtools$ head -n 3 cpg.bed exons.bed
==> cpg.bed <==
chr1 28735 29810 CpG:_116
chr1 135124 135563 CpG:_30
chr1 327790 328229 CpG:_29
==> exons.bed <==
chr1 11873 12227 NR_046018_exon_0_0_chr1_11874_f 0 +
chr1 12612 12721 NR_046018_exon_1_0_chr1_12613_f 0 +
chr1 13220 14409 NR_046018_exon_2_0_chr1_13221_f 0 +获得重叠区域(既是外显子,又是CpG岛的区域)
ct@ehbio:~/bedtools$ bedtools intersect -a cpg.bed -b exons.bed | head -5
chr1 29320 29370 CpG:_116
chr1 135124 135563 CpG:_30
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29
chr1 327790 328229 CpG:_29输出重叠区域对应的原始区域(与外显子存在交集的CpG岛)
ct@ehbio:~/bedtools$ bedtools intersect -a cpg.bed -b exons.bed -wa -wb > | head -5- chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r 0 -
- chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 -
- chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 +
- chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 +
- chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 +
计算重叠碱基数
ct@ehbio:~/bedtools$ bedtools intersect -a cpg.bed -b exons.bed -wo | head -10- chr1 28735 29810 CpG:_116 chr1 29320 29370 NR_024540_exon_10_0_chr1_29321_r 0 - 50
- chr1 135124 135563 CpG:_30 chr1 134772 139696 NR_039983_exon_0_0_chr1_134773_r 0 - 439
- chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028322_exon_2_0_chr1_324439_f 0 + 439
- chr1 327790 328229 CpG:_29 chr1 324438 328581 NR_028325_exon_2_0_chr1_324439_f 0 + 439
- chr1 327790 328229 CpG:_29 chr1 327035 328581 NR_028327_exon_3_0_chr1_327036_f 0 + 439
- chr1 713984 714547 CpG:_60 chr1 713663 714068 NR_033908_exon_6_0_chr1_713664_r 0 - 84
- chr1 762416 763445 CpG:_115 chr1 761585 762902 NR_024321_exon_0_0_chr1_761586_r 0 - 486
- chr1 762416 763445 CpG:_115 chr1 762970 763155 NR_015368_exon_0_0_chr1_762971_f 0 + 185
- chr1 762416 763445 CpG:_115 chr1 762970 763155 NR_047519_exon_0_0_chr1_762971_f 0 + 185
- chr1 762416 763445 CpG:_115 chr1 762970 763155 NR_047520_exon_0_0_chr1_762971_f 0 + 185
计算第一个(-a)bed区域有多少个重叠的第二个(-b)bed文件中有多少个区域
ct@ehbio:~/bedtools$ bedtools intersect -a cpg.bed -b exons.bed -c | head
chr1 28735 29810 CpG:_116 1
chr1 135124 135563 CpG:_30 1
chr1 327790 328229 CpG:_29 3
chr1 437151 438164 CpG:_84 0
chr1 449273 450544 CpG:_99 0
chr1 533219 534114 CpG:_94 0
chr1 544738 546649 CpG:_171 0
chr1 713984 714547 CpG:_60 1
chr1 762416 763445 CpG:_115 10
chr1 788863 789211 CpG:_28 9另外还有-v取出不重叠的区域, -f限定重叠最小比例,-sorted可以对按sort -k1,1 -k2,2n排序好的文件加速操作。
同时对多个区域求交集 (可以用于peak的多维注释)
# -names标注注释来源
# -sorted: 如果使用了这个参数,提供的一定是排序好的bed文件
ct@ehbio:~/bedtools$ bedtools intersect -a exons.bed \
-b cpg.bed gwas.bed hesc.chromHmm.bed -sorted -wa -wb -names cpg gwas chromhmm \
| head -10000 | tail -10- chr1 27632676 27635124 NM_001276252_exon_15_0_chr1_27632677_chromhmm chr1 27633213 27635013 5_Strong_Enhancer
- chr1 27632676 27635124 NM_001276252_exon_15_0_chr1_27632677_chromhmm chr1 27635013 27635413 7_Weak_Enhancer
- chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f chromhmm chr1 27632613 27632813 6_Weak_Enhancer
- chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f chromhmm chr1 27632813 27633213 7_Weak_Enhancer
- chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f chromhmm chr1 27633213 27635013 5_Strong_Enhancer
- chr1 27632676 27635124 NM_015023_exon_15_0_chr1_27632677_f chromhmm chr1 27635013 27635413 7_Weak_Enhancer
- chr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f cpg chr1 27648453 27649006 CpG:_63
- chr1 27648635 27648882 NM_032125_exon_0_0_chr1_27648636_f chromhmm chr1 27648613 27649413 1_Active_Promoter
- chr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f cpg chr1 27648453 27649006 CpG:_63
- chr1 27648635 27648882 NR_037576_exon_0_0_chr1_27648636_f chromhmm chr1 27648613 27649413 1_Active_Promoter
- 合并区域
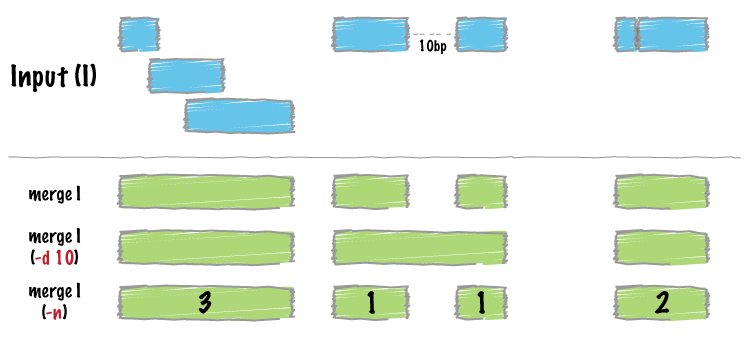
Figure 5.2: bedtools merge
bedtools merge输入的是按sort -k1,1 -k2,2n排序好的bed文件。
只需要输入一个排序好的bed文件,默认合并重叠或邻接区域。
ct@ehbio:~/bedtools$ bedtools merge -i exons.bed | head -n 5
chr1 11873 12227
chr1 12612 12721
chr1 13220 14829
chr1 14969 15038
chr1 15795 15947合并区域并输出此合并后区域是由几个区域合并来的
ct@ehbio:~/bedtools$ bedtools merge -i exons.bed -c 1 -o count | head -n 5
chr1 11873 12227 1
chr1 12612 12721 1
chr1 13220 14829 2
chr1 14969 15038 1
chr1 15795 15947 1合并相距90 nt内的区域,并输出是由哪些区域合并来的
# -c: 指定对哪些列进行操作
# -o: 与-c对应,表示对指定列进行哪些操作
# 这里的用法是对第一列做计数操作,输出这个区域是由几个区域合并来的
# 对第4列做收集操作,记录合并的区域的名字,并逗号分隔显示出来
ct@ehbio:~/bedtools$ bedtools merge -i exons.bed -d 340 -c 1,4 -o count,collapse | head -4
chr1 11873 12227 1 NR_046018_exon_0_0_chr1_11874_f
chr1 12612 12721 1 NR_046018_exon_1_0_chr1_12613_f
chr1 13220 15038 3 NR_046018_exon_2_0_chr1_13221_f,NR_024540_exon_0_0_chr1_14362_r,NR_024540_exon_1_0_chr1_14970_r
chr1 15795 15947 1 NR_024540_exon_2_0_chr1_15796_r- 计算互补区域
给定一个全集,再给定一个子集,求另一个子集。比如给定每条染色体长度和外显子区域,求非外显子区域。给定基因区,求非基因区。给定重复序列,求非重复序列等。
重复序列区域的获取也可以用上面提供的链接 http://blog.genesino.com/2013/05/ucsc-usages/。
ct@ehbio:~/bedtools$ head genome.txt
chr1 249250621
chr10 135534747
chr11 135006516
chr11_gl000202_random 40103
chr12 133851895
chr13 115169878
chr14 107349540
chr15 102531392
ct@ehbio:~/bedtools$ bedtools complement -i exons.bed -g genome.txt | head -n 5
chr1 0 11873
chr1 12227 12612
chr1 12721 13220
chr1 14829 14969
chr1 15038 15795- 基因组覆盖广度和深度
计算基因组某个区域是否被覆盖,覆盖深度多少。有下图多种输出格式,也支持RNA-seq数据,计算junction-reads覆盖。
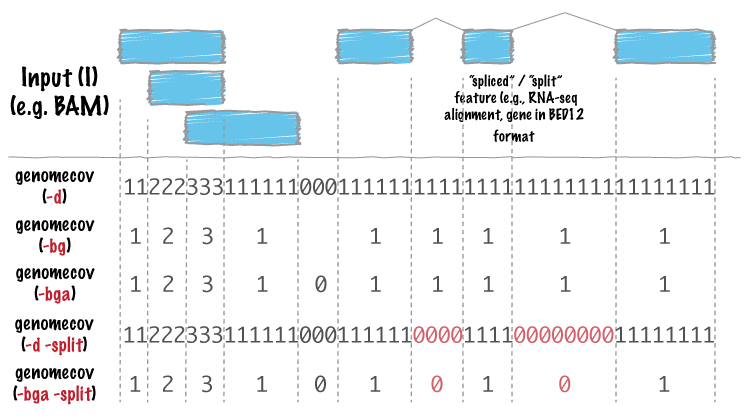
Figure 5.3: bedtools genomecov
genome.txt里面的内容就是染色体及对应的长度。
# 对单行FASTA,可如此计算
# 如果是多行FASTA,则需要累加
ct@ehbio:~/bedtools$ awk 'BEGIN{OFS=FS="\t"}{\
if($0~/>/) {seq_name=$0;sub(">","",seq_name);} \
else {print seq_name,length;} }' ../bio/genome.fa | tee ../bio/genome.txt
chr1 60001
chr2 54001
chr3 54001
chr4 60001
ct@ehbio:~/bedtools$ bedtools genomecov -ibam ../bio/map.sortP.bam -bga \
-g ../bio/genome.txt | head
# 这个warning很有意思,因为BAM中已经有这个信息了,就不需要提供了
*****
*****WARNING: Genome (-g) files are ignored when BAM input is provided.
*****
# bedgraph文件,前3列与bed相同,最后一列表示前3列指定的区域的覆盖度。
chr1 0 11 0
chr1 11 17 1
chr1 17 20 2
chr1 20 31 3
chr1 31 36 4
chr1 36 43 6
chr1 43 44 7
chr1 44 46 8
chr1 46 48 9
chr1 48 54 10两个思考题:
- 怎么计算有多少基因组区域被测到了?
- 怎么计算平均测序深度是多少?
- 数据集相似性
bedtools jaccard计算的是给定的两个bed文件之间交集区域(intersection)占总区域(union-intersection)的比例(jaccard)和交集的数目(n_intersections)。
ct@ehbio:~/bedtools$ bedtools jaccard \
-a fHeart-DS16621.hotspot.twopass.fdr0.05.merge.bed \
-b fHeart-DS15839.hotspot.twopass.fdr0.05.merge.bed
intersection union-intersection jaccard n_intersections
81269248 160493950 0.50637 130852小思考:1. 如何用bedtools其它工具算出这个结果?2. 如果需要比较的文件很多,怎么充分利用计算资源?
一个办法是使用for循环, 双层嵌套。这种用法也很常见,不管是单层还是双层for循环,都有利于简化重复运算。
ct@ehbio:~/bedtools$ for i in *.merge.bed; do \
for j in *.merge.bed; do \
bedtools jaccard -a $i -b $j | cut -f3 | tail -n +2 | sed "s/^/$i\t$j\t/"; \
done; done >total.similarity另一个办法是用parallel,不只可以批量,更可以并行。
root@ehbio:~# yum install parallel.noarch# parallel 后面双引号("")内的内容为希望用parallel执行的命令,
# 整体写法与Linux下命令写法一致。
# 双引号后面的 三个相邻冒号 (:::)默认用来传递参数的,可多个连写。
# 每个三冒号后面的参数会被循环调用,而在命令中的引用则是根据其出现的位置,分别用{1}, {2}
# 表示第一个三冒号后的参数,第二个三冒号后的参数。
#
# 这个命令可以替换原文档里面的整合和替换, 相比于原文命令生成多个文件,这里对每个输出结果
# 先进行了比对信息的增加,最后结果可以输入一个文件中。
#
ct@ehbio:~/bedtools$ parallel "bedtools jaccard -a {1} -b {2} | awk 'NR> | cut -f 3 \
| sed 's/^/{1}\t{2}\t/'" ::: `ls *.merge.bed` ::: `ls *.merge.bed` >totalSimilarity.2
# 上面的命令也有个小隐患,并行计算时的输出冲突问题,可以修改为输出到单个文件,再cat到一起
ct@ehbio:~/bedtools$ parallel "bedtools jaccard -a {1} -b {2} | awk 'NR> | cut -f 3 \
| sed 's/^/{1}\t{2}\t/' >{1}.{2}.totalSimilarity_tmp" ::: `ls *.merge.bed` ::: `ls *.merge.bed`
ct@ehbio:~/bedtools$ cat *.totalSimilarity_tmp >totalSimilarity.2
# 替换掉无关信息
ct@ehbio:~/bedtools$ sed -i -e 's/.hotspot.twopass.fdr0.05.merge.bed//' \
-e 's/.hg19//' totalSimilarity.2 原文档的命令,稍微有些复杂,利于学习不同命令的组合。使用时推荐使用上面的命令。
ct@ehbio:~/bedtools$ parallel "bedtools jaccard -a {1} -b {2} \
| awk 'NR>1' | cut -f 3 \
> {1}.{2}.jaccard" \
::: `ls *.merge.bed` ::: `ls *.merge.bed`This command will create a single file containing the pairwise Jaccard measurements from all 400 tests.
find . \
| grep jaccard \
| xargs grep "" \
| sed -e s"/\.\///" \
| perl -pi -e "s/.bed./.bed\t/" \
| perl -pi -e "s/.jaccard:/\t/" \
> pairwise.dnase.txtA bit of cleanup to use more intelligible names for each of the samples.
cat pairwise.dnase.txt \
| sed -e 's/.hotspot.twopass.fdr0.05.merge.bed//g' \
| sed -e 's/.hg19//g' \
> pairwise.dnase.shortnames.txtNow let’s make a 20x20 matrix of the Jaccard statistic. This will allow the data to play nicely with R.
awk 'NF==3' pairwise.dnase.shortnames.txt \
| awk '$1 ~ /^f/ && $2 ~ /^f/' \
| python make-matrix.py \
> dnase.shortnames.distance.matrix5.7 SRA toolkit使用
SRA toolkit https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software根据服务器下载对应的二进制编码包。
CentOS下地址:https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.9.0/sratoolkit.2.9.0-centos_linux64.tar.gz。
常用的是fastq-dump,从NCBI的SRA数据库下载测序原始文件并转化为FASTQ格式供后续分析使用。
# --split-3: 若是双端测序则拆分
# --gzip: 拆分后压缩文件
# fastq-dump -v --split-3 --gzip SRR_number
fastq-dump -v --split-3 --gzip SRR502564下面是我写的一个脚本,有3个用途。一是在fastq-dump下载过程中,如果断了可以再次启动下载;二是下载完成之后,给下载的文件进行重命名为方便识别的名字;三是清空下载缓存。
所需要的输入文件是一个2列文件,第一列为SRR号,第二列为样品名字,TAB键分割。
SRR2952884 Y25_root
SRR2952883 Y18_root
SRR2952882 Y12_root
SRR2952881 Y05_root#!/bin/bash
set -x
set -e
set -u
usage()
{
cat <<EOF >&2
${txtcyn}
Usage:
$0 options${txtrst}
${bldblu}Function${txtrst}:
This script is used to download sra files from a file containing SRA
accession numbers and transfer SRA to fastq format.
The format of input file:
SRR2952884 Y25_root
SRR2952883 Y18_root
SRR2952882 Y12_root
SRR2952881 Y05_root
The second column will be treated as the prefix of final fastq files.
${txtbld}OPTIONS${txtrst}:
-f Data file with format described above${bldred}[NECESSARY]${txtrst}
-z Is there a header[${bldred}Default TRUE${txtrst}]
EOF
}
file=
while getopts "hf:z:" OPTION
do
case $OPTION in
h)
usage
exit 1
;;
f)
file=$OPTARG
;;
?)
usage
exit 1
;;
esac
done
if [ -z $file ]; then
usage
exit 1
fi
#IFS="\t"
cat $file | while read -r -a array
do
sra="${array[0]}"
name="${array[1]}"
#echo $sra, $name
#prefetch -v $sra
#prefetch -v $sra
#prefetch -v $sra
#/bin/cp ~/ncbi/public/sra/${sra}.sra ${name}.sra
while true
do
fastq-dump -v --split-3 --gzip ${sra}
a=$?
if [ "$a" == "0" ]; then break; fi
sleep 5m
done
#/bin/cp ~/ncbi/public/sra/${sra}* .
if [ "$a" == "0" ]
then
rename "${sra}" "${name}" ${sra}*
/bin/rm ~/ncbi/public/sra/${sra}.sra
fi
done5.8 生信流程开发
最基本的是Bash脚本,把上面call SNP的命令放到一个Bash脚本文件中即可。另外可以使用Makefile和Airflow进行更高级一些的开发。
Airflow使用见 http://blog.genesino.com/2016/05/airflow/。
一篇不错的英文Makefile教程 http://blog.genesino.com/2011/04/introduction-to-making-makefiles/。
5.9 数据同步和备份
5.9.1 原创拷贝scp
最简单的备份方式,就是使用cp (本地硬盘)或 scp (远程硬盘)命令,给自己的结果文件新建一个拷贝;每有更新,再拷贝一份。具体命令如下:
cp -fur source_project project_bak
scp -r source_project user@remote_server_ip:project_bak为了实现定期备份,我们可以把上述命令写入crontab程序中,设置每天的晚上23:00执行。对于远程服务器的备份,我们可以配置免密码登录,便于自动备份。后台输入免密码登录服务器,获取免密码登录服务器的方法。
# Crontab format
# Minute Hour Day Month Week command
# * 表示每分/时/天/月/周
# 每天23:00 执行cp命令
0 23 * * * cp -fur source_project project_bak
# */2 表示每隔2分分/时/天/月/周执行命令
# 每隔24小时执行cp命令
0 */24 * * * cp -fur source_project project_bak
0 0 */1 * * scp -r source_project user@remote_server_ip:project_bak
# 另外crotab还有个特殊的时间
# @reboot: 开机运行指定命令
@reboot cmd5.9.2 镜像备份和增量同步 rsync
cp或scp使用简单,但每次执行都会对所有文件进行拷贝,耗时耗力,尤其是需要拷贝的内容很多时,重复拷贝对时间和硬盘都是个损耗。
rsync则是一个增量备份工具,只针对修改过的文件的修改过的部分进行同步备份,大大缩短了传输的文件的数量和传输时间。具体使用如下 :
# 把本地project目录下的东西备份到远程服务器的/backup/project目录下
# 注意第一个project后面的反斜线,表示拷贝目录内的内容,不在目标目录新建project文件夹。注意与第二个命令的比较,两者实现同样的功能。
# -a: archive mode, quals -rlptgoD
# -r: 递归同步
# -p: 同步时保留原文件的权限设置
# -u: 若文件在远端做过更新,则不同步,避免覆盖远端的修改
# -L: 同步符号链接链接的文件,防止在远程服务器出现文件路径等不匹配导致的软连接失效
# -t: 保留修改时间
# -v: 显示更新信息
# -z: 传输过程中压缩文件,对于传输速度慢时适用
rsync -aruLptvz --delete project/ user@remoteServer:/backup/project
rsync -aruLptvz --delete project user@remoteServer:/backup/rsync所做的工作为镜像,保证远端服务器与本地文件的统一。如果本地文件没问题,远端也不会有问题。但如果发生误删或因程序运行错误,导致文件出问题,而在同步之前又没有意识到的话,远端的备份也就没了备份的意义,因为它也被损坏了。误删是比较容易发现的,可以及时矫正。但程序运行出问题,则不一定了。
5.9.3 增量备份,记录各个版本 rdiff-backup
这里推荐一个工具rdiff-backup不只可以做增量备份,而且会保留每次备份的状态,新备份和上一次备份的差别,可以轻松回到之前的某个版本。唯一的要求就是,本地服务器和远端服务器需要安装统一版本的rdiff-backup。另外还有2款工具 duplicity和`Rsnapshot也可以做类似工作,但方法不一样,占用的磁盘空间也不一样,具体可查看原文链接中的比较。
具体的rdiff-backup安装和使用见http://mp.weixin.qq.com/s/c2cspK5b4sQScWYMBtG63g。
5.10 References
- 高通量数据分析必备-基因组浏览器使用介绍 - 1
- 高通量数据分析必备-基因组浏览器使用介绍 - 2
- 高通量数据分析必备-基因组浏览器使用介绍 - 3
- 测序文章数据上传找哪里
- GO、GSEA富集分析一网打进
- GSEA富集分析 - 界面操作
- Bedtools使用简介
- OrthoMCL鉴定物种同源基因 (安装+使用)
- Rfam 12.0+本地使用 (最新版教程)
- 轻松绘制各种Venn图
- ETE构建、绘制进化树
- psRobot:植物小RNA分析系统
- 生信软件系列 - NCBI使用
- 去东方,最好用的在线GO富集分析工具
- 2018 升级版Motif数据库Jaspar
- 一文教会你查找基因的启动子、UTR、TSS等区域以及预测转录因子结合位点